





When preparing molecular dynamics simulations, one often encounters the concept of periodic boundary conditions (PBC). While PBC allows molecules to interact as if they were in an infinite system (by replicating the simulation box in all directions), there’s a crucial rule that must be adhered to for the simulation to produce accurate results: the minimum image convention. So what exactly does this mean for your molecular modeling? Let’s break it down!
What is the Minimum Image Convention?
In molecular simulations using PBC, each particle only interacts with the closest periodic image of every other particle. This principle ensures that short-range, non-bonded interactions such as van der Waals or electrostatic forces are computed efficiently and accurately.
However, a key point to remember is that if particles in the system are too close to the box boundaries, they might interact with their own periodic images. This can distort the forces and lead to inaccurate simulation results.
To avoid this issue, the simulation box should always provide sufficient distance between solute molecules and the box’s boundary.
How to Satisfy the Minimum Image Convention
A general rule of thumb to satisfy this convention is:
Leave at least
1.0 nmbetween the solute and the box boundary. This ensures at least2.0 nmof space between periodic images of the solute.
By following this guideline, molecular interactions become more physically accurate, and simulation artifacts are minimized.
Practical Tool: GROMACS Wizard in SAMSON
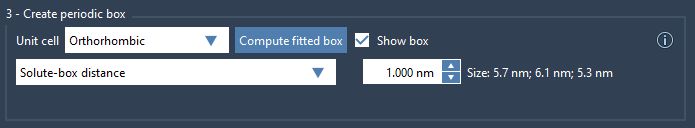
In SAMSON’s GROMACS Wizard, you have access to tools for setting up the simulation box and ensuring compliance with the minimum image convention. Two options are provided to help you tailor the box to your system:
- Box Lengths: You can directly specify the box’s dimensions. When fitted tightly to your system, you may need to manually increase the size to maintain the required distance between periodic images.
- Solute-Box Distance: This option lets you define the distance between your system (solute) and the box. A recommended value of at least
1.0 nmensures compliance with the minimum image convention and provides the flexibility to handle batch projects or path simulations.
Why is Box Shape Important?
The shape of the simulation box also plays a role in its efficiency. For approximately spherical molecules, such as some proteins, non-cubic unit cells like rhombic dodecahedrons or truncated octahedrons are favored. These shapes minimize the number of solvent molecules required to surround the solute while still satisfying the minimum image constraints. For instance:
- Rhombic Dodecahedron: This shape saves up to 29% CPU time since its volume is only 71% that of a cube with the same image distance.
Here’s an example of how you can adjust the box in GROMACS Wizard:
Additional Considerations
It’s also worth noting that while box shapes in SAMSON can be modified, GROMACS employs a brick-shaped volume (for computational efficiency) during actual simulations. SAMSON can detect the unit cell shape automatically when you load GROMACS trajectories, allowing you to refine the system further if needed.
Interested in mastering periodic boundary conditions and unit cell shapes further? Explore the detailed explanations at the GROMACS Wizard Documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. Get SAMSON today at SAMSON Connect.