





For molecular modelers working on viral proteins, a key challenge often lies in understanding the motion and dynamics of key components of the virus. The SARS-CoV-2 spike protein – the mechanism the virus uses to enter human cells – is an intriguing example. A deep understanding of its opening and closing motion is critical for designing effective antiviral strategies, particularly when it comes to targeting the receptor-binding domain (RBD).
This article explores the process of modeling the motion of the SARS-CoV-2 spike protein. By observing its transition from a closed state to an open state, insights can be gained into how the spike binds to the ACE2 receptor on human cells. Here, we provide a walkthrough of this motion, along with details on how it was computed using molecular design tools in SAMSON.
Why the Spike Motion Matters
Understanding the spike’s opening motion matters because this transition enables the virus to recognize and bind to human cells. The spike consists of three S-proteins forming a trimer, and the receptor-binding domain (RBD) at the tip of one of these proteins swings open to bind to the ACE2 receptor on host cells.
The spike motion also explains why its upper region, exposed for receptor binding, becomes the primary target for neutralizing antibodies. These insights are valuable for vaccine design, antiviral drug discovery, and the development of therapeutic antibodies.
Visualizing the Spike in Action
Animations of the spike’s motion provide a clear perspective on its behavior. The three animations below depict the spike from different angles, revealing its dynamic transition from a closed (down) state to an open (up) state, where it can bind ACE2:
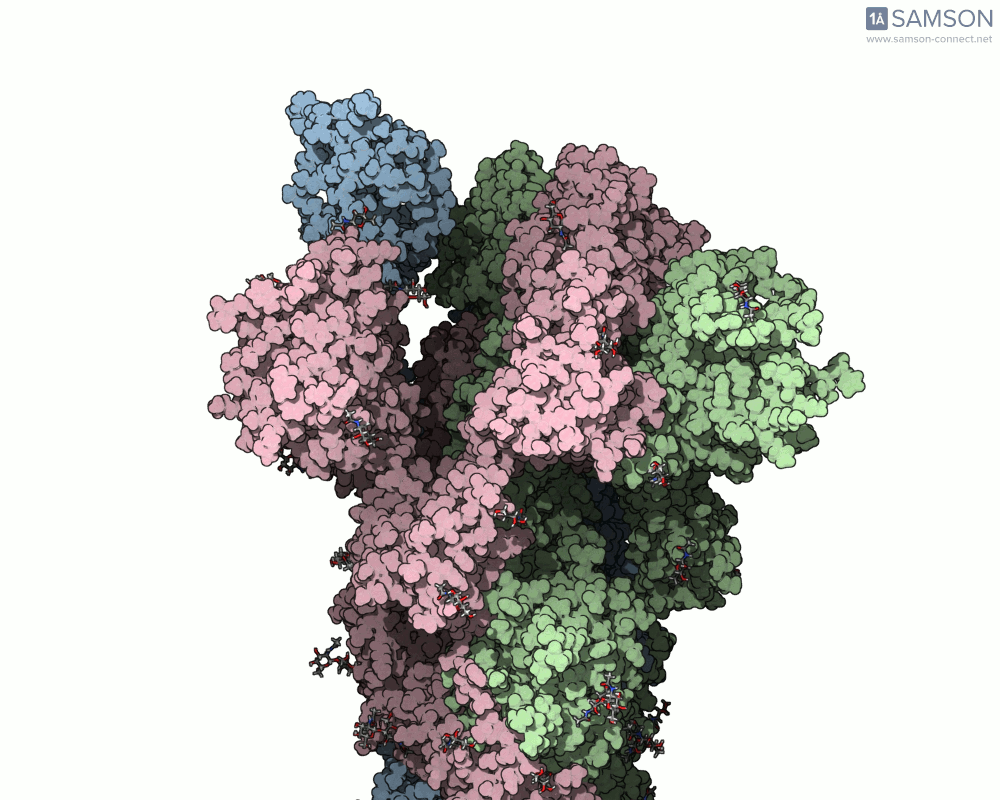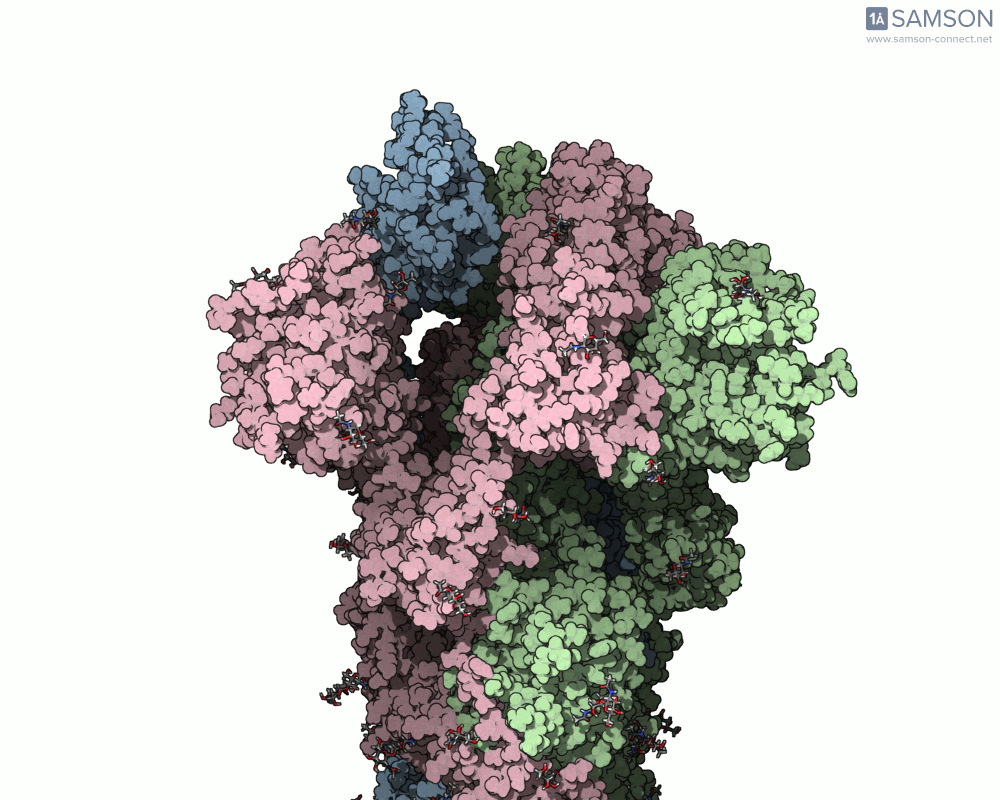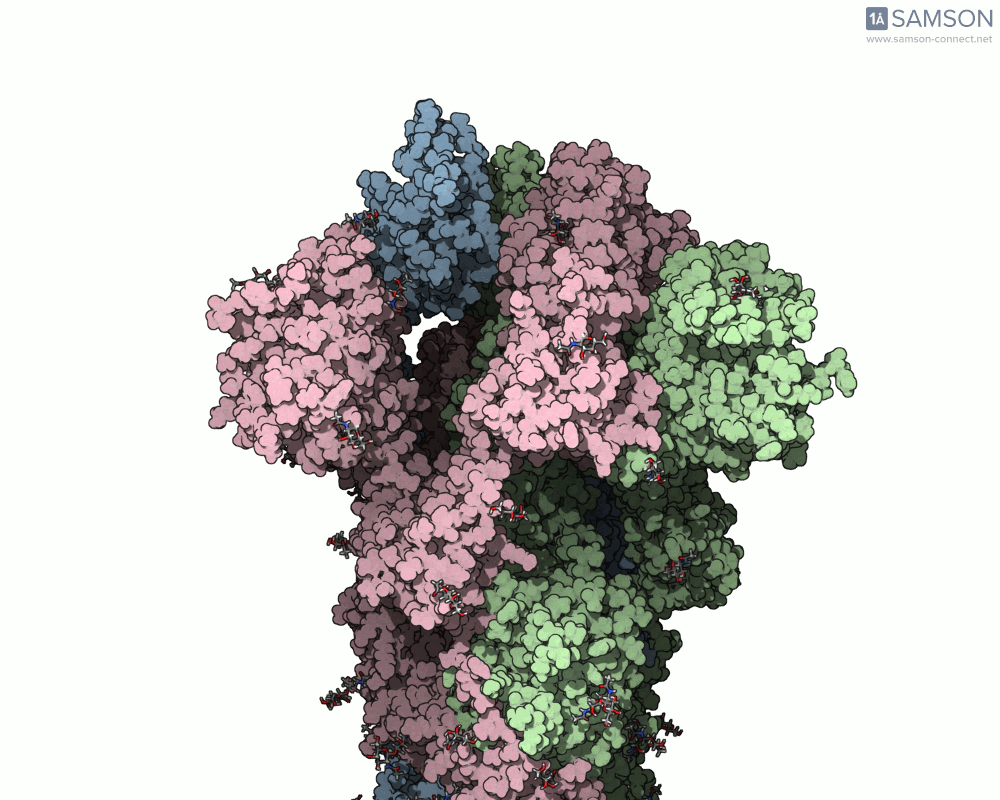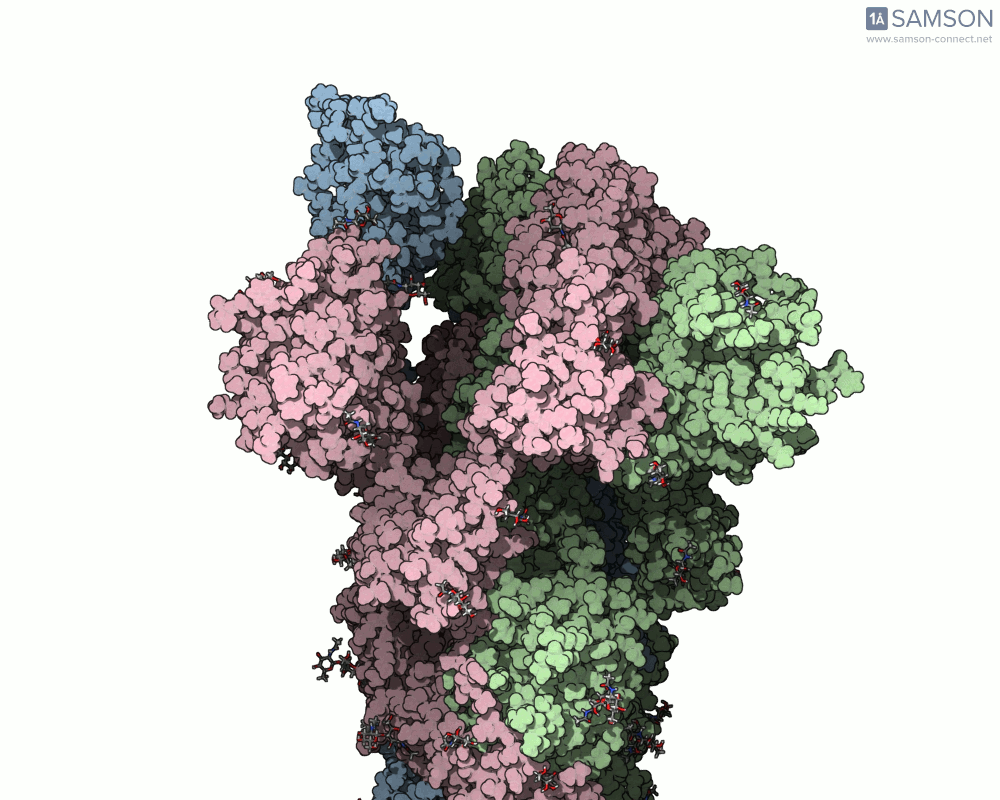
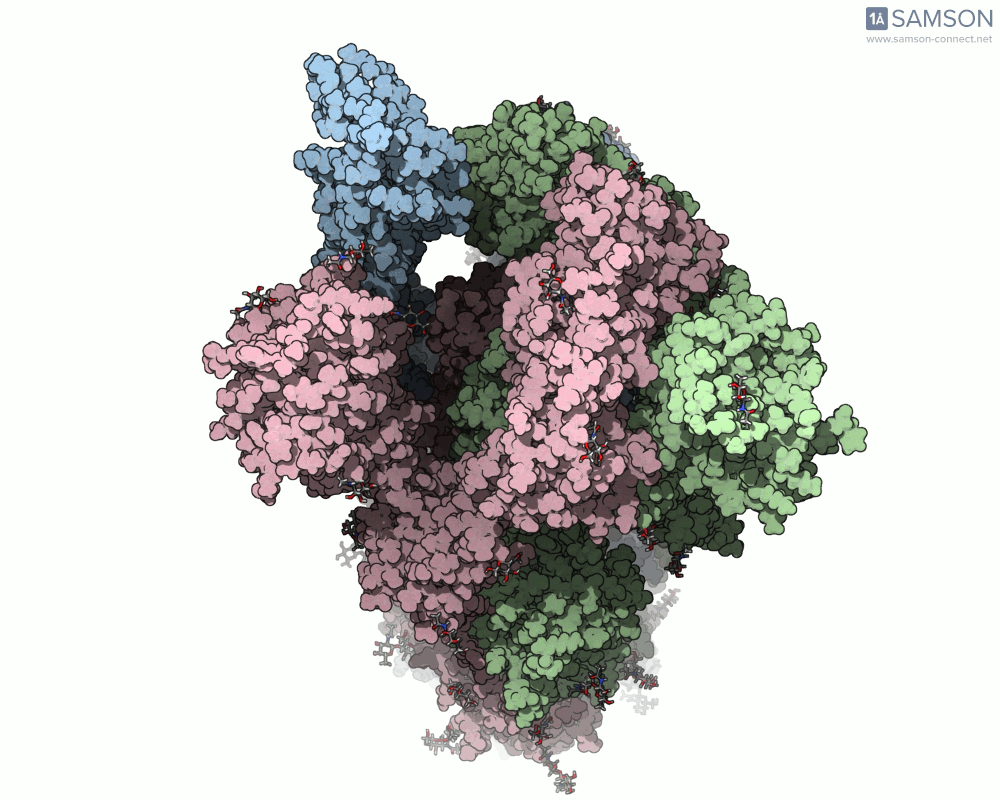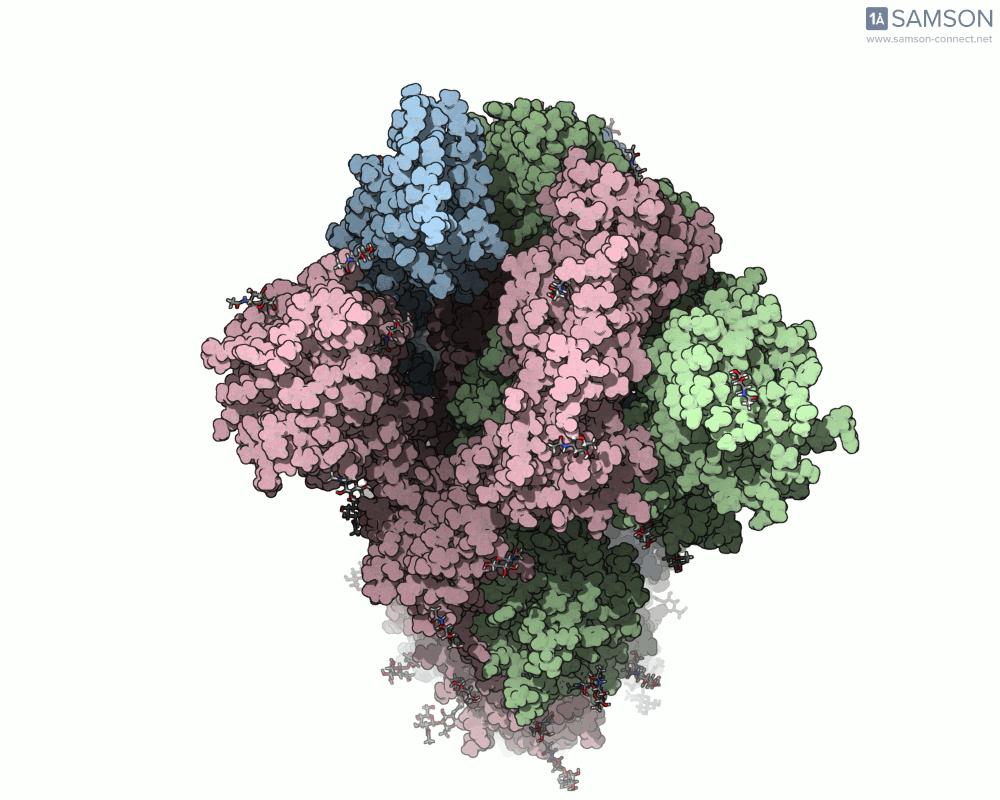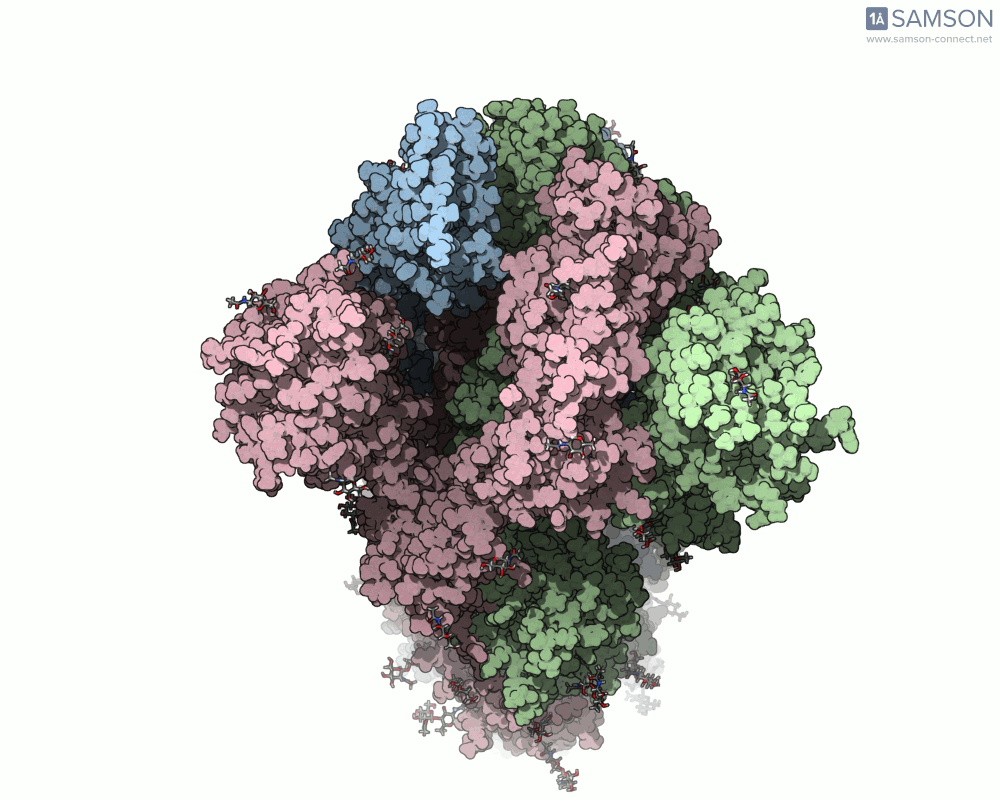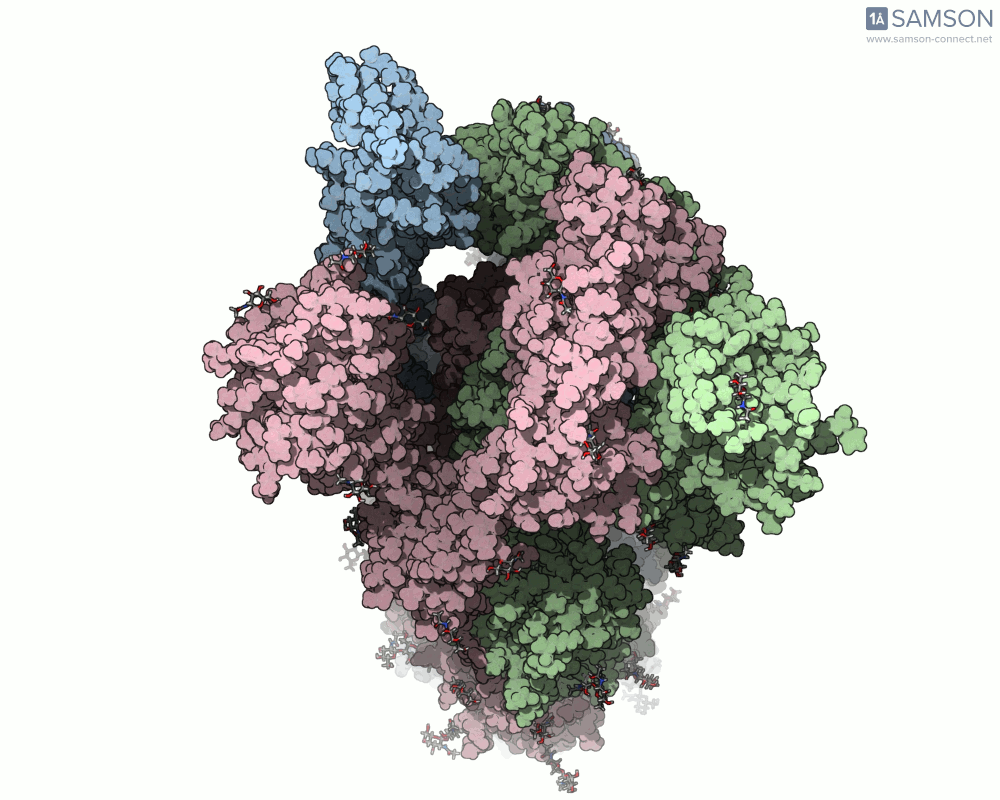
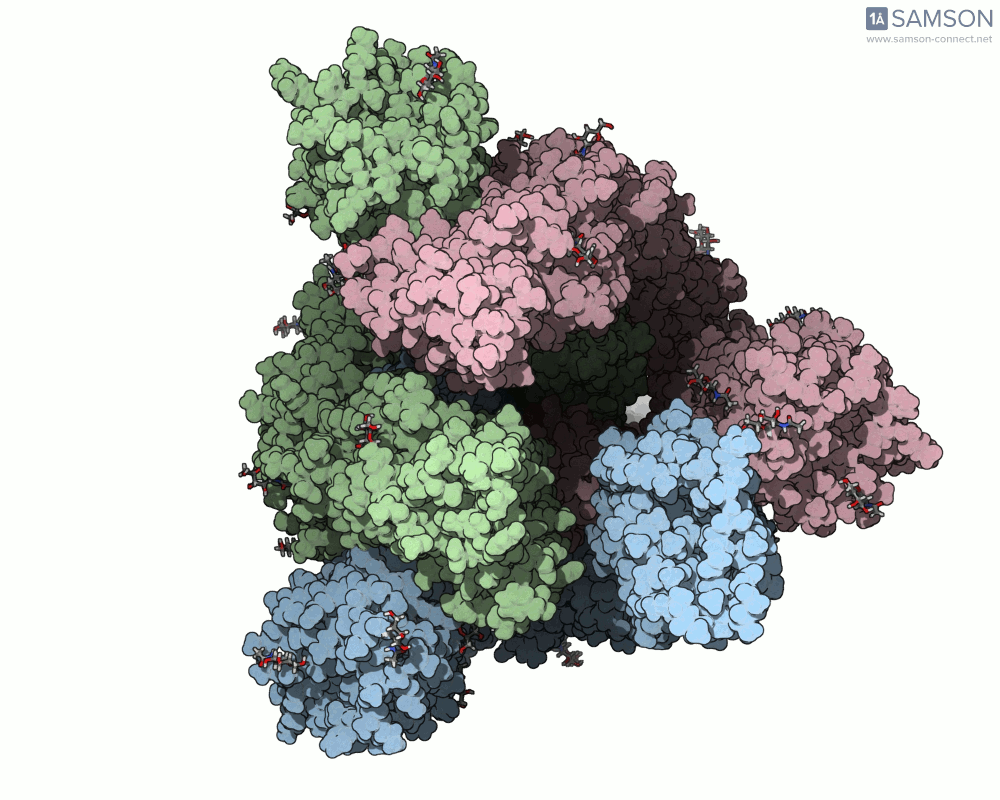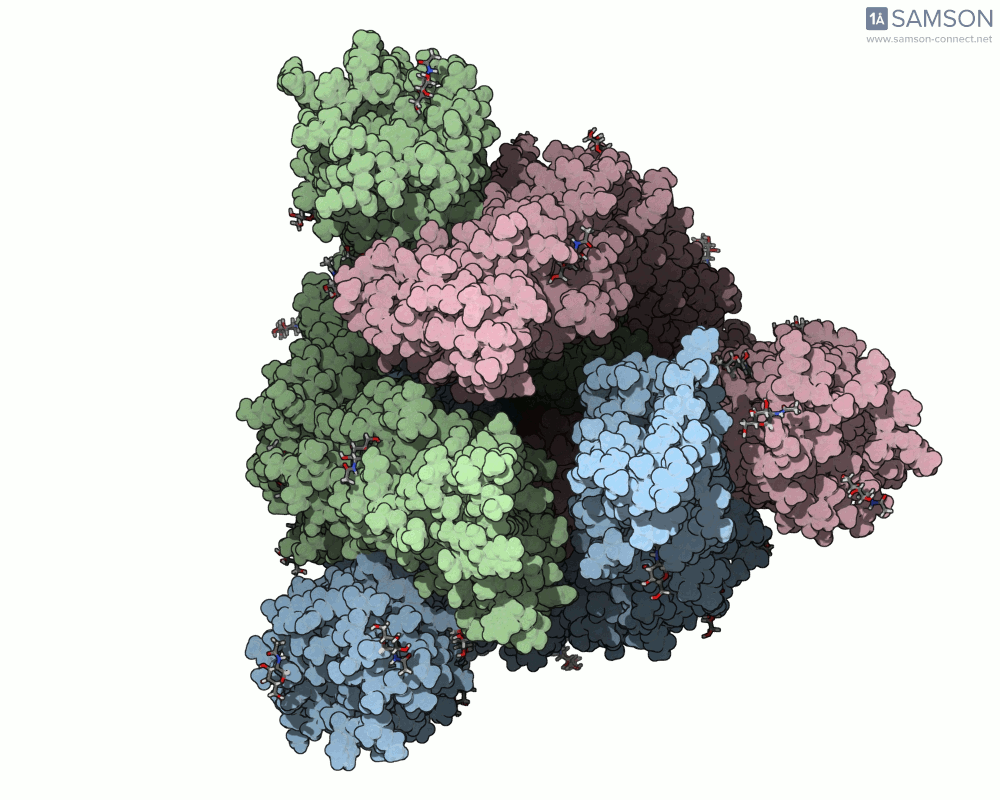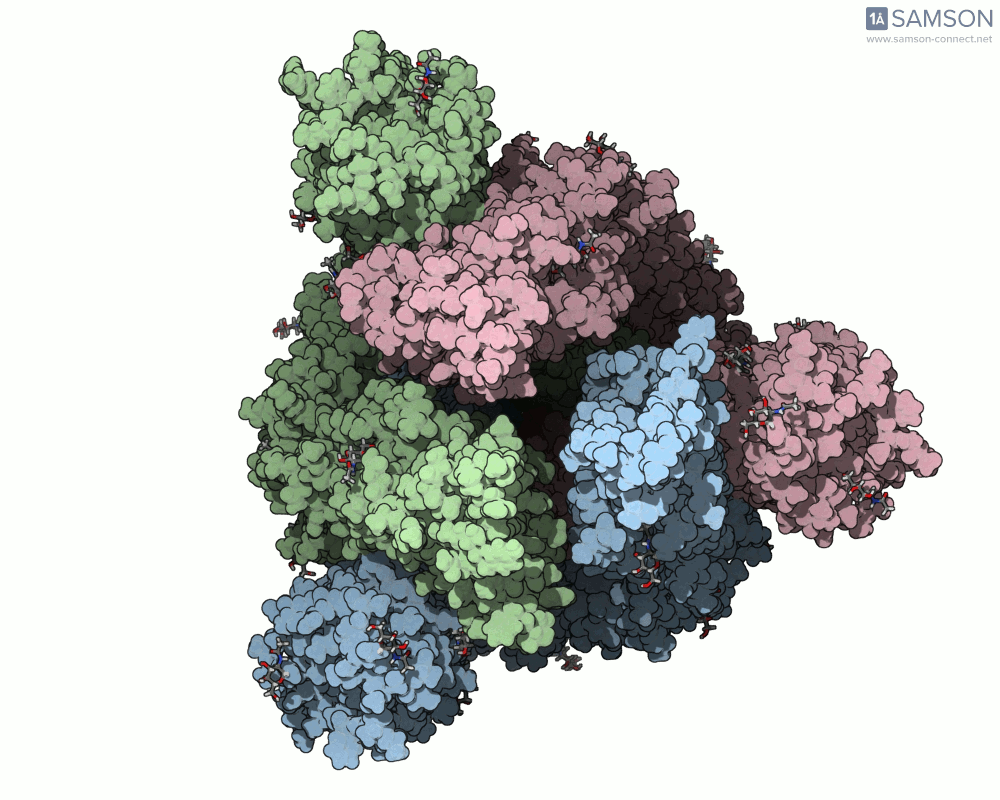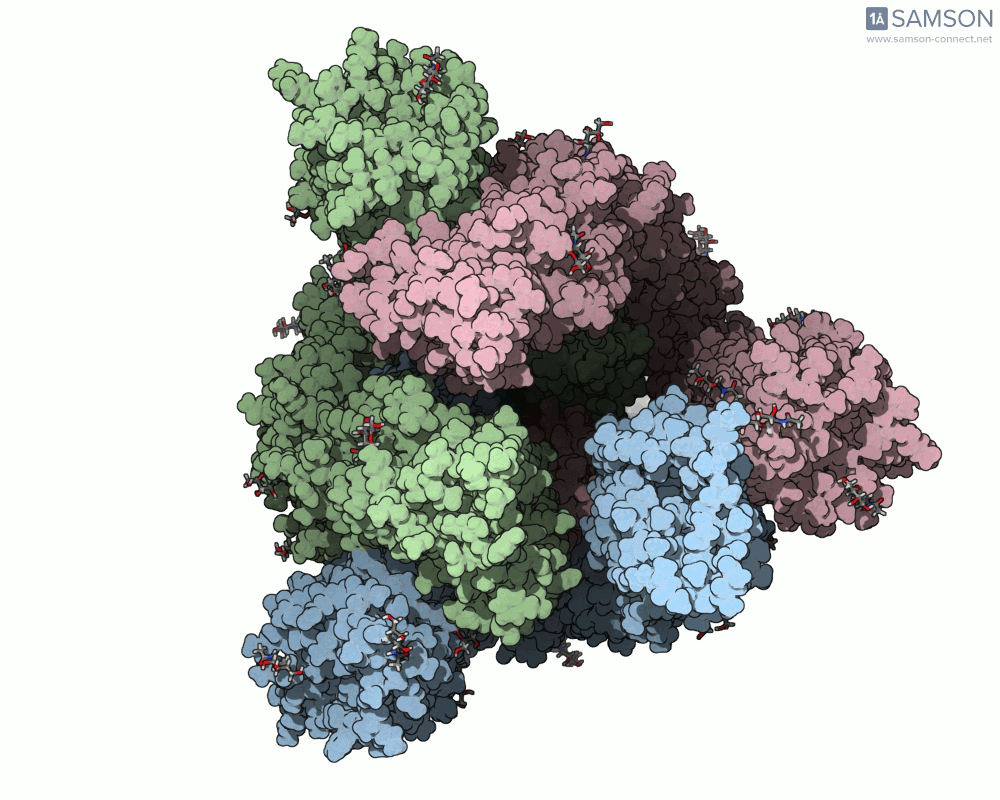
These visualizations bring to life the mechanics of viral entry, which can be pivotal in subsequent simulations and computational studies.
Accessing the Computed Trajectories
SAMSON users can download the computed trajectory files used in this visualization. The different formats available include:
These files are useful for modelers who want to analyze the simulated results or integrate them into their own workflows. The SAMSON file seamlessly incorporates features such as double-click animations and trajectory paths computed using advanced SAMSON modules.
How the Motion Was Computed in SAMSON
The spike’s motion was initialized using two known conformations of the protein: the closed state (PDB 6VXX) and the open state (PDB 6VYB). While the differing number of residues between these two structures made the process challenging, a systematic pipeline in SAMSON ensured accurate interpolation:
- Bond orders in sugars were set using a Python script to prepare for hydrogen addition.
- The structures were minimized after hydrogens were added.
- The ARAP Interpolation Path module was used to generate an initial path between the open and closed conformations in only seconds.
- Further refinement was achieved using the P-NEB module, which optimized the transition path in around 15 minutes.
This procedure highlights how SAMSON enables researchers to create actionable molecular motion paths quickly and efficiently, even when the structural models differ.
To learn more about the SARS-CoV-2 spike and explore hands-on resources, visit the full documentation here.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.