





For molecular modelers diving into the intricacies of SARS-CoV-2 research, understanding the spike protein’s structural dynamics is crucial. The transition of the spike from its closed state to its open state is particularly significant, as this conformational shift enables the virus to recognize and bind to ACE2 receptors on human cells. Yet, visualizing these motions can feel daunting without the right tools or data. Fortunately, SAMSON provides a pathway to compute, visualize, and explore this motion efficiently.
Let’s delve into how this motion was computed, observed, and how you can use it to advance your molecular modeling projects.
The Challenge: Modeling Structural Transitions
One common struggle for modelers is bridging the gap between two experimentally determined conformations of a molecule. In the case of the SARS-CoV-2 spike, researchers had access to two critical structures:
- Closed state: PDB 6VXX
- Open state: PDB 6VYB
However, these structures differed in the number of residues, increasing the complexity of modeling a smooth transition between them. This is where SAMSON’s robust modules, such as the ARAP (As-Rigid-As-Possible) Interpolation Path and Parallel Nudged Elastic Band (P-NEB), played an essential role in refining the computed motion.
Computing the Motion
The development of the spike motion pipeline involved the following main steps:
- Adjusting the bond orders in sugar residues using a Python script, ensuring correct preparation for hydrogen addition and further steps.
- Adding hydrogens to both structures (closed and open) and carrying out energy minimization to refine them.
- Generating a preliminary interpolated path using the ARAP module, starting from the open state (6VYB) and targeting the closed state (6VXX). This initial step provided an overview of how the spike might move structurally.
- Refining the path with the P-NEB module, which optimized the trajectory transitions between key conformations and accounted for residue-based differences.
Thanks to the efficiency of SAMSON, the ARAP step required less than a minute on a laptop, while the more involved P-NEB refinement took about 15 minutes. This demonstrates how computational tools can save hours or even days of work for molecular modelers.
See It In Motion
Below are illustrative visualizations of the spike protein transitioning from its closed to open state. These views highlight the spike’s capacity to recognize ACE2 receptors, underscoring its relevance in facilitating viral infection:
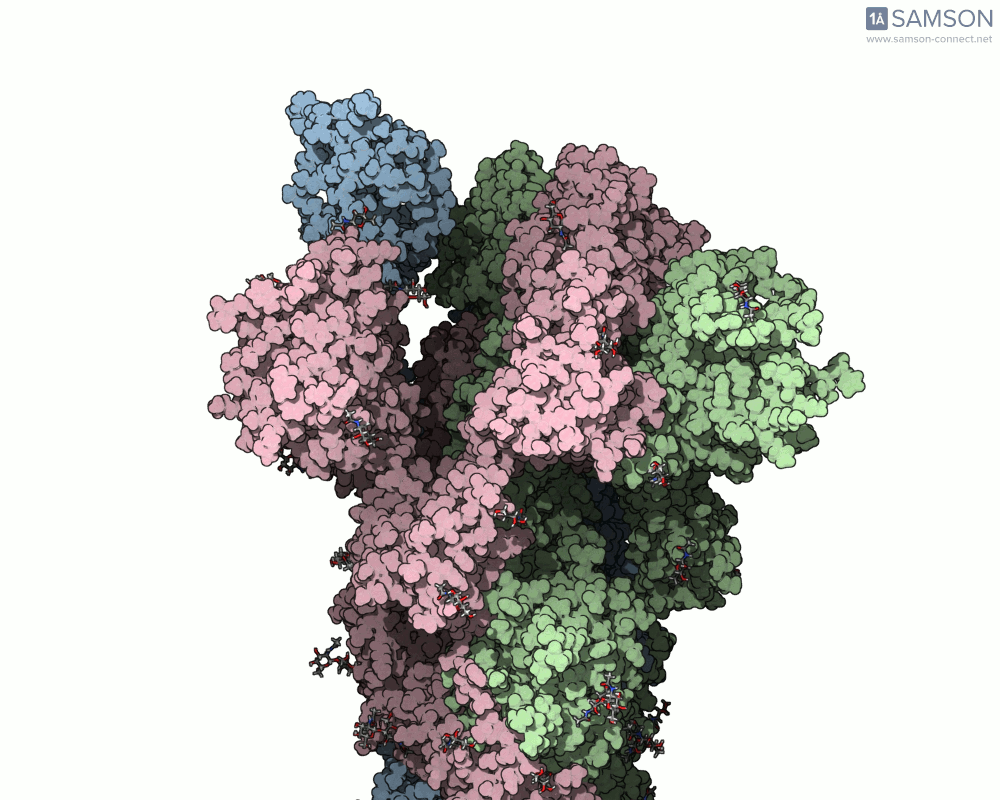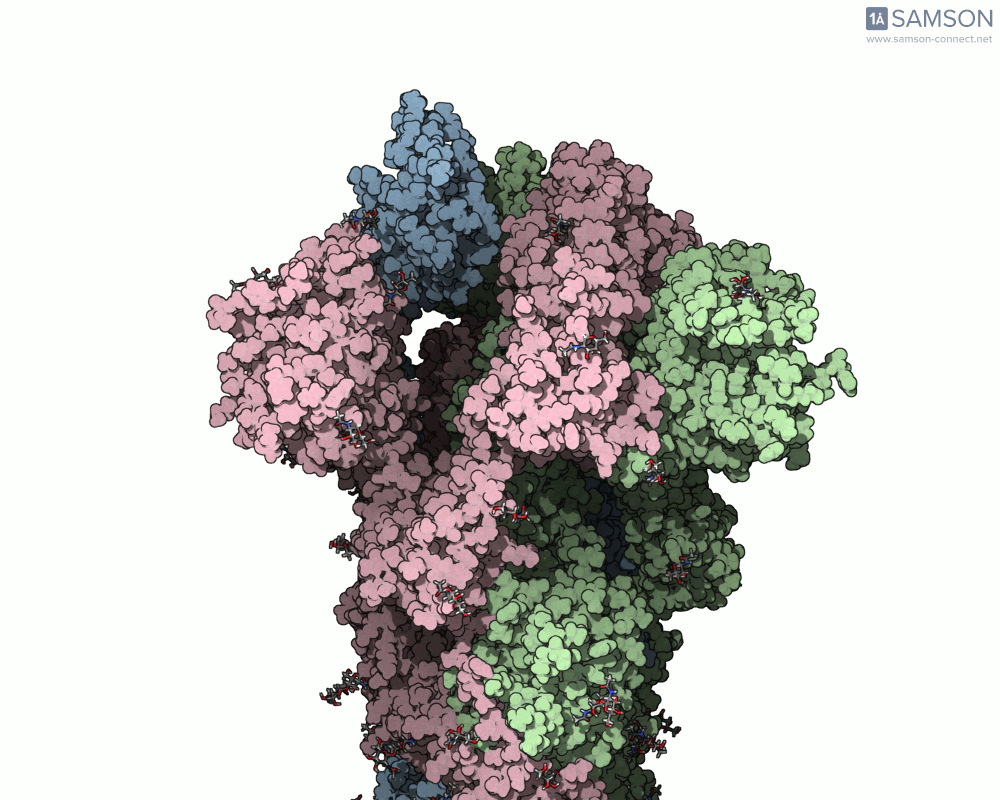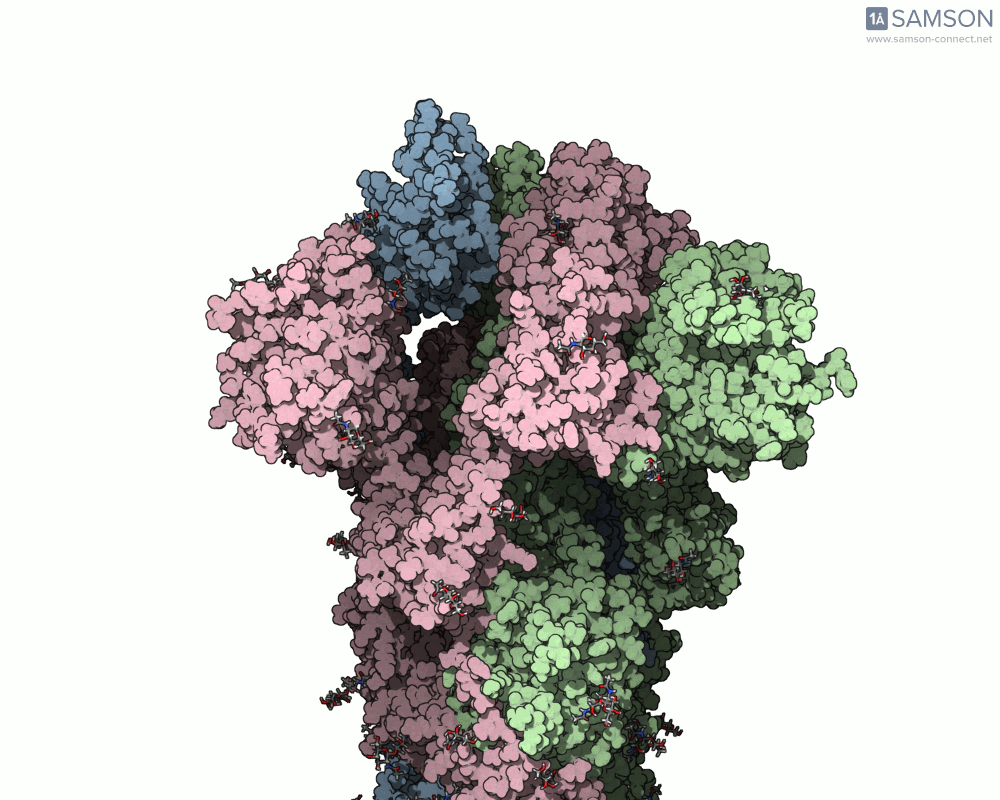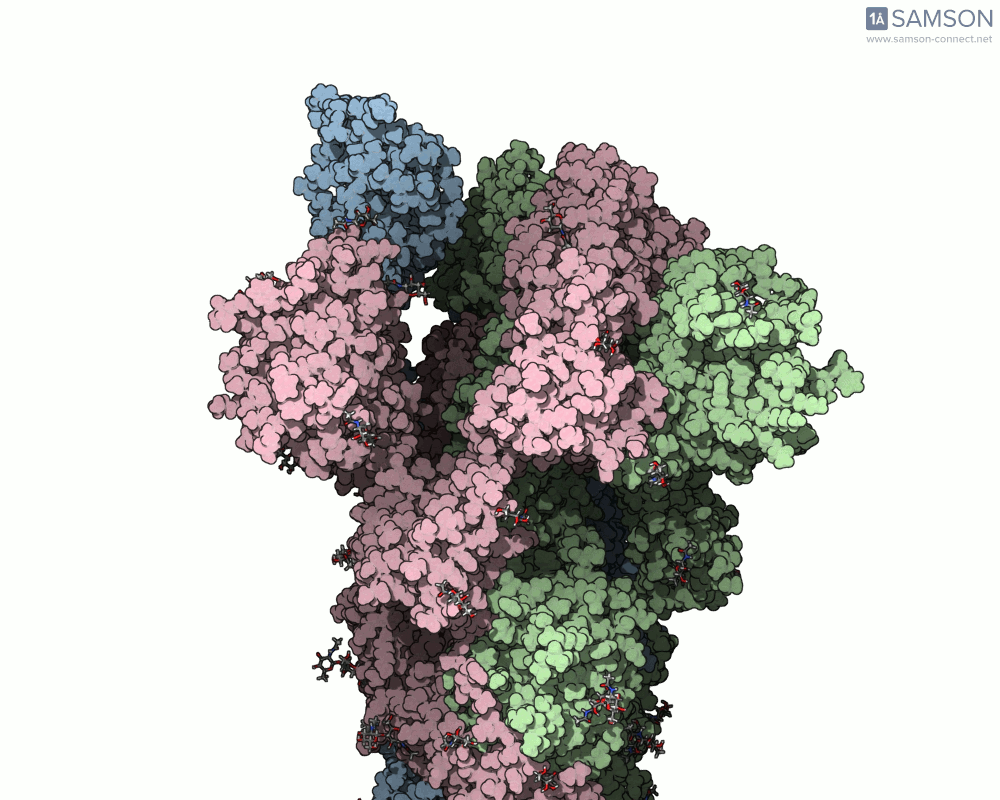
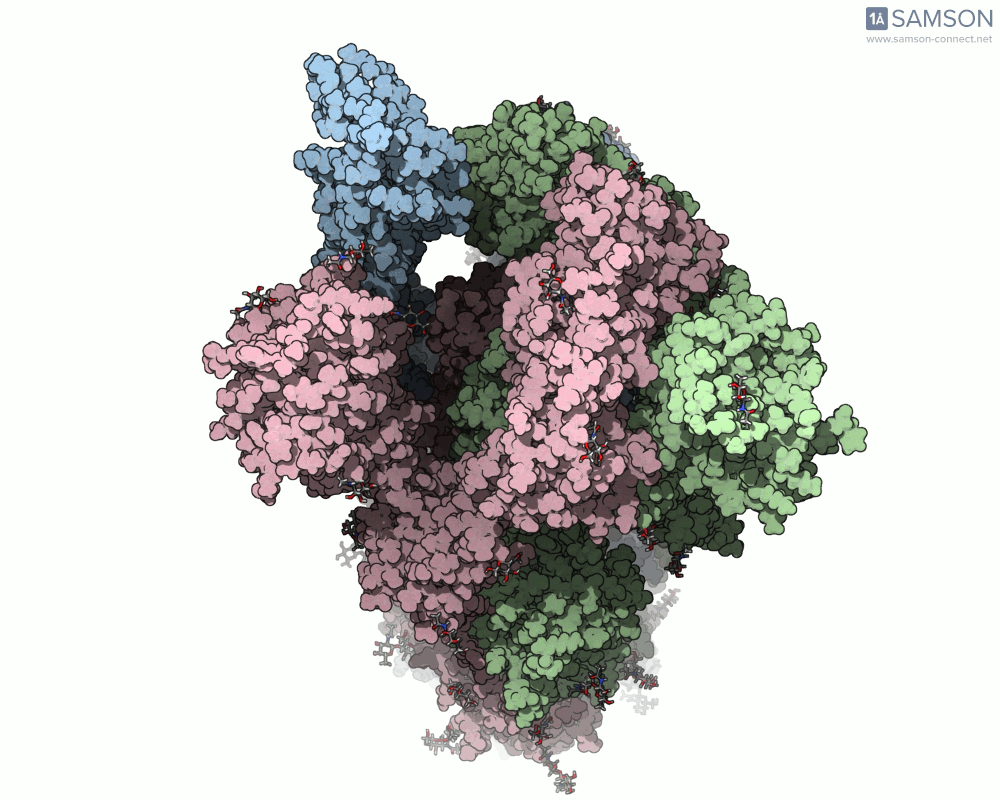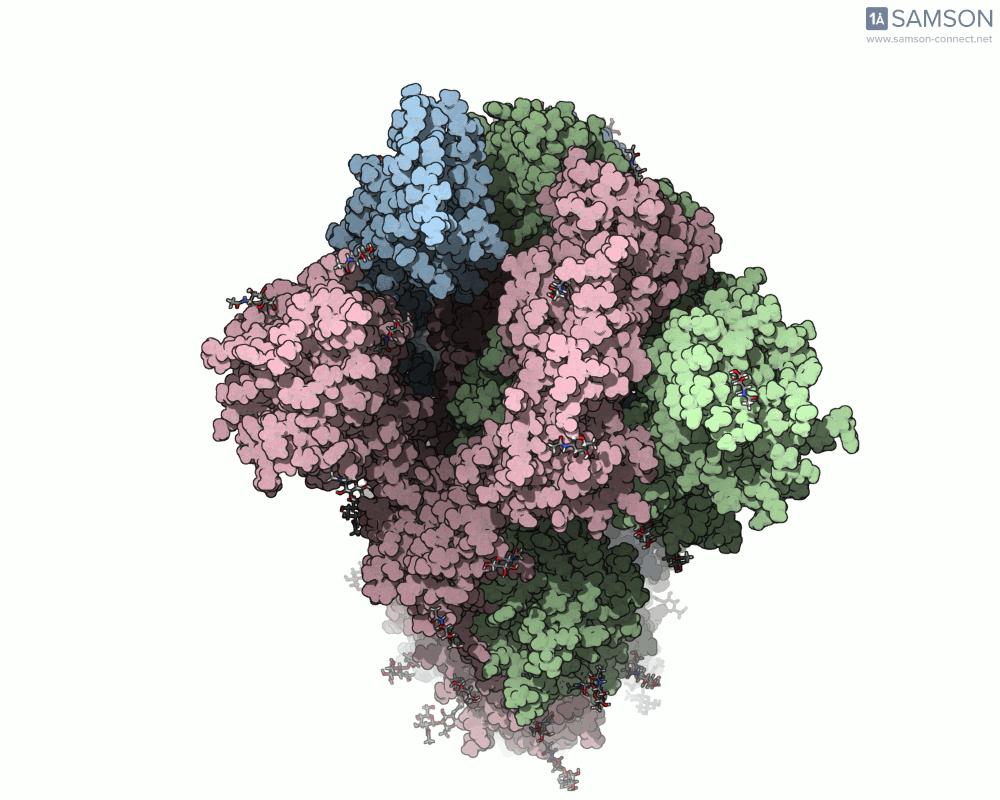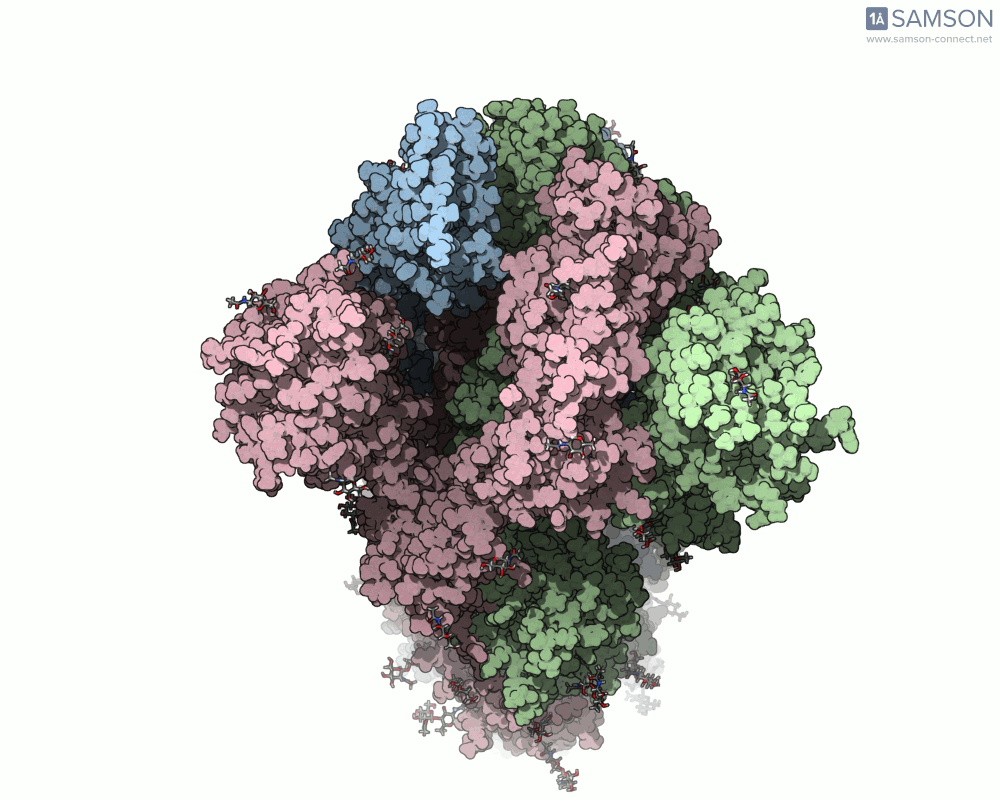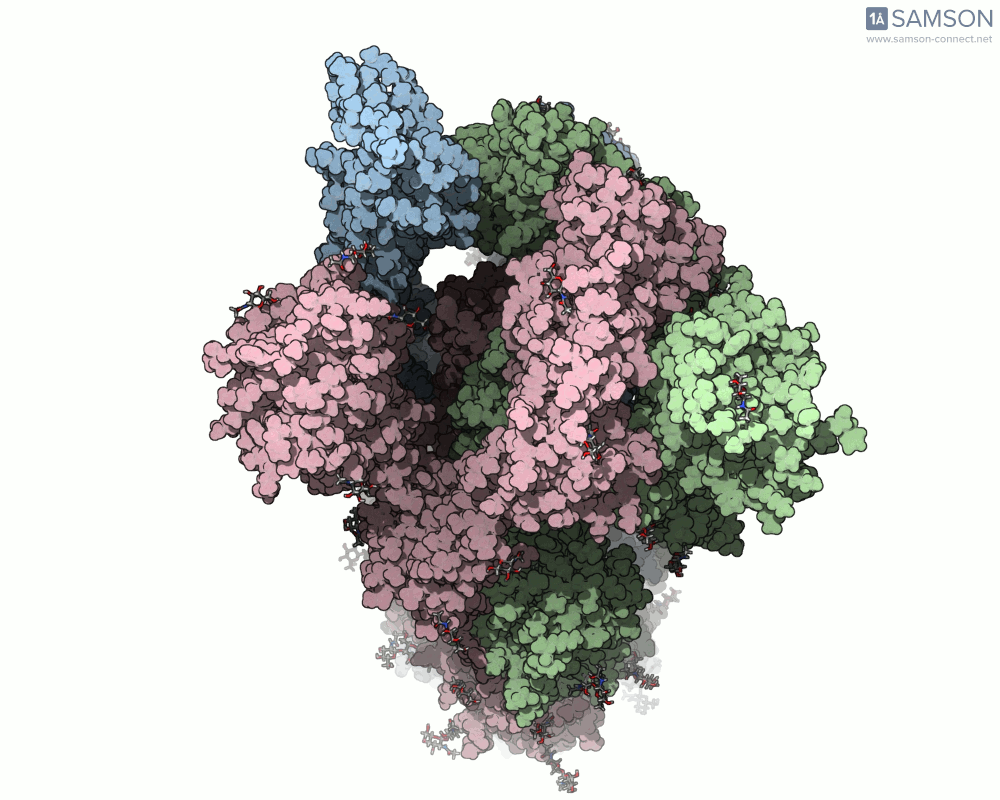
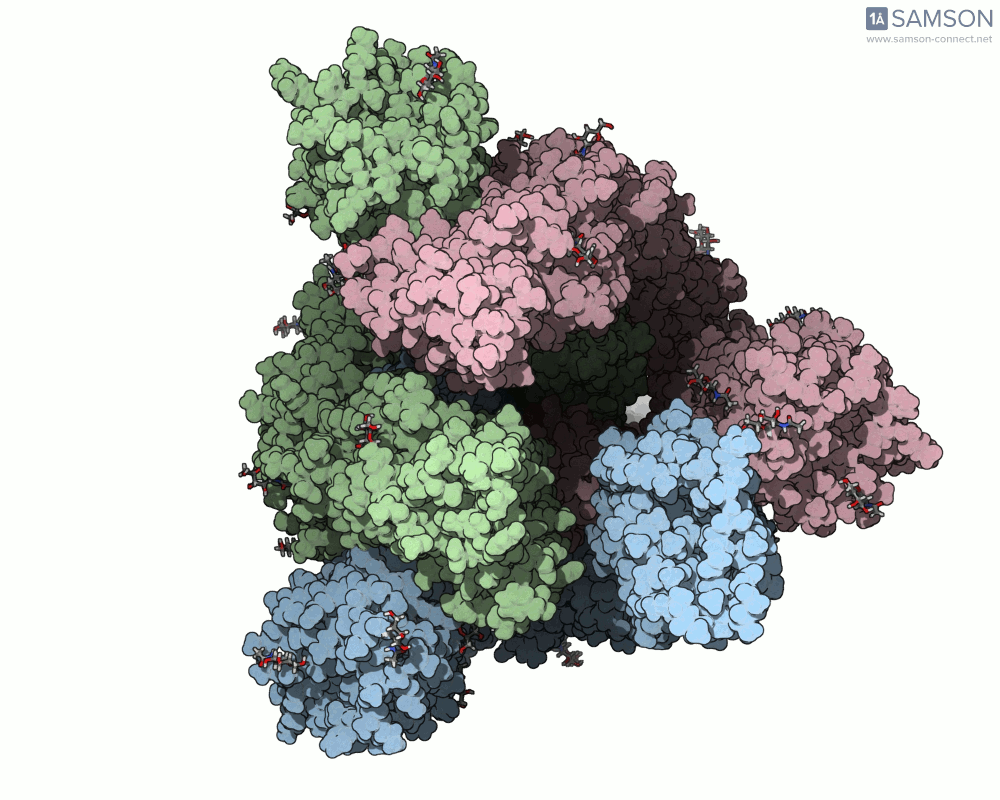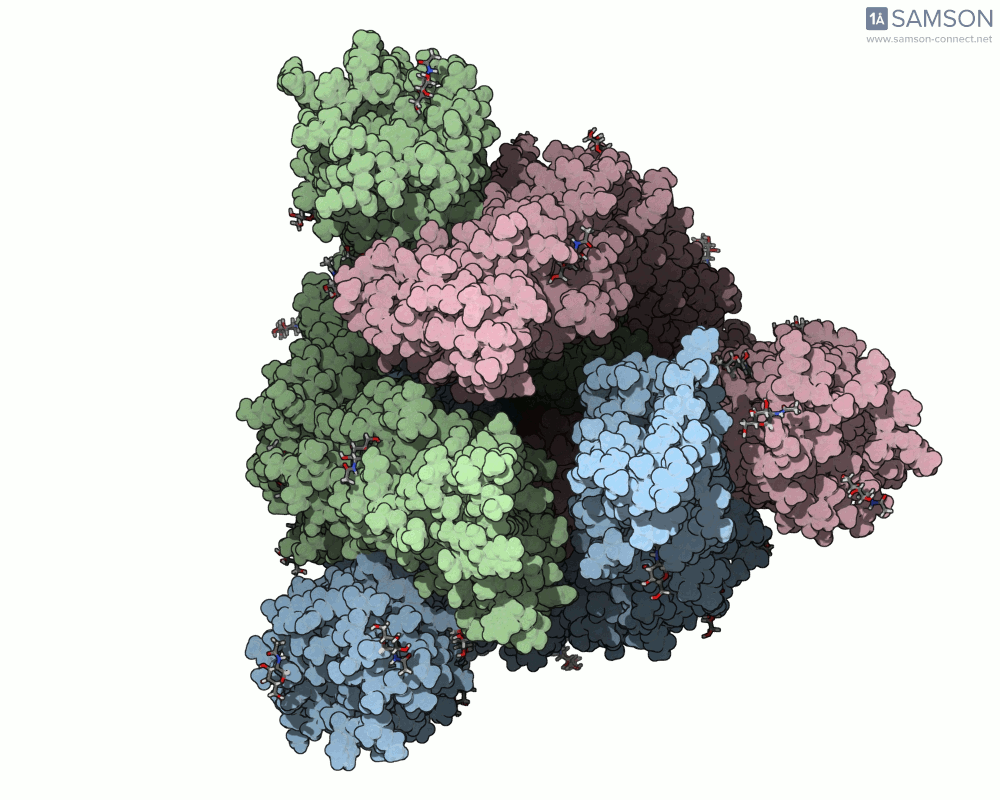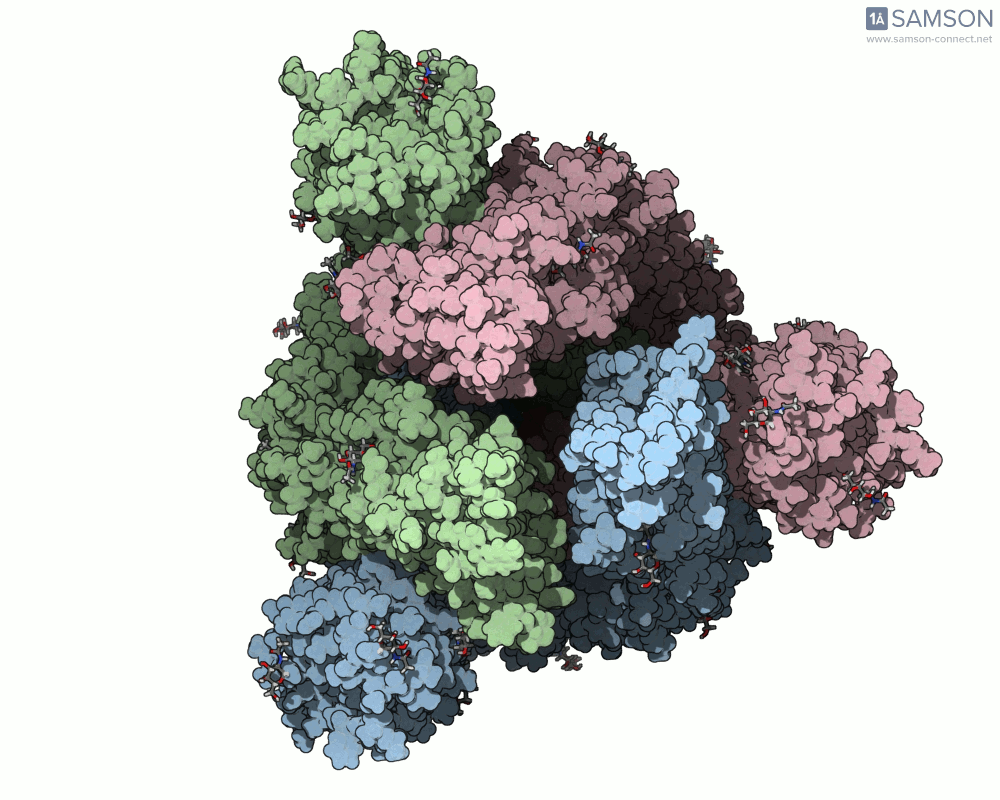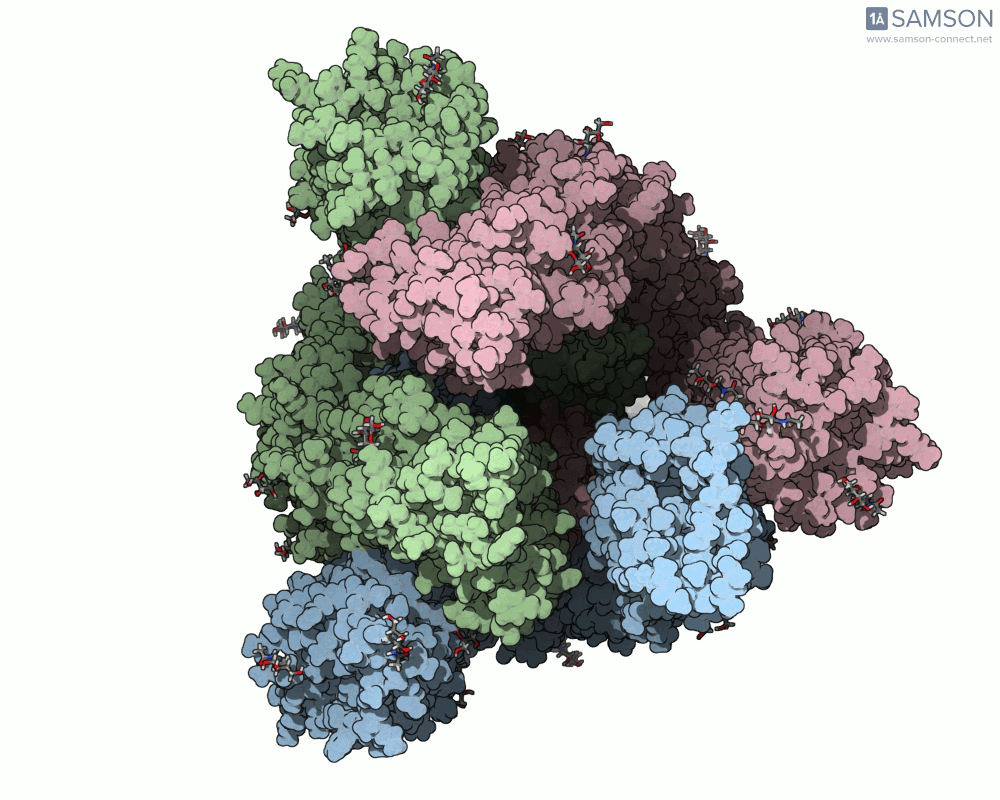
The trajectory files used for these animations are available for download in multiple formats (PDB and SAMSON format), allowing molecular modelers to conduct further analysis or even post-process the data.
How You Can Apply This
If you’re designing antibodies, inhibitors, or modeling binding interactions, these trajectory files and computed pathways offer an invaluable resource. Knowing how the protein transitions between states can shed light on intermediate conformations, potentially revealing transient binding sites or interactions.
For more technical details on the pipeline and access to module links, visit the original documentation page at SAMSON’s SARS-CoV-2 Spike Protein Documentation.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. Get started today by downloading SAMSON at https://www.samson-connect.net.