





For molecular modelers, creating realistic and biologically meaningful transition paths between protein conformations can be challenging and time-consuming. Whether you’re working on conformational analysis, transition state modeling, or setting up an umbrella sampling workflow, an efficient and precise interpolation method can make all the difference. This guide introduces you to the ARAP (As-Rigid-As-Possible) Interpolation Extension in SAMSON, a tool that allows you to generate smooth, consistent transition paths in seconds.
Why ARAP Interpolation?
Traditional methods for modeling intermediate states between protein conformations can result in unrealistic shifts or artifacts that don’t align with biological relevance. The ARAP Interpolation Extension circumvents this by leveraging a geometrically meaningful model that treats the structure as rigid as possible, while still accommodating necessary adjustments. Here’s why you might consider it:
- Compute continuous structural transitions in seconds, saving valuable time.
- Visualize and export intermediate conformations with ease.
- Generate paths that align with biologically meaningful geometries, ensuring realistic reaction coordinates.
- Compatible with downstream workflows, such as free energy simulations or structural ensemble visualizations.
Step-by-Step: From Protein Structures to Transition Path
Let’s dive into creating a transition path for the Diphtheria Toxin protein between two conformations: 1DDT and 1MDT.
1. Prepare Your Structures
Fetch and clean your base structures directly within SAMSON:
- Go to Home > Fetch, and input
1DDT 1MDTunder PDB or PDB (mmCIF). - Load the structures and focus on the
Achain of each by deleting theBchain from1MDT. - Clean the structures via Home > Prepare to remove unnecessary elements like water, ions, and ligands.
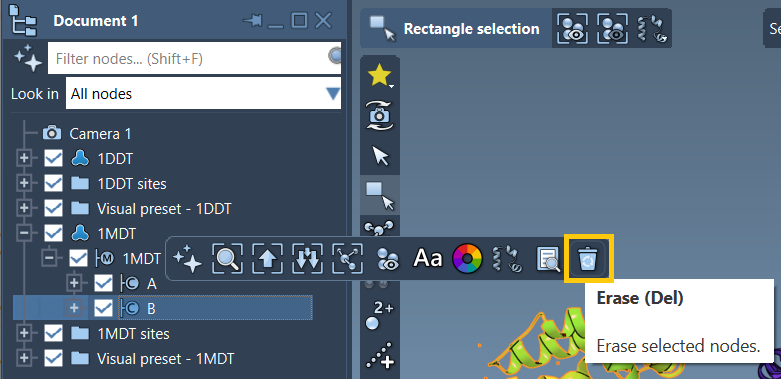
Your goal: Navigate from chain A of 1DDT to chain A of 1MDT.
2. Create Conformations
Next, create conformations from the cleaned structures:
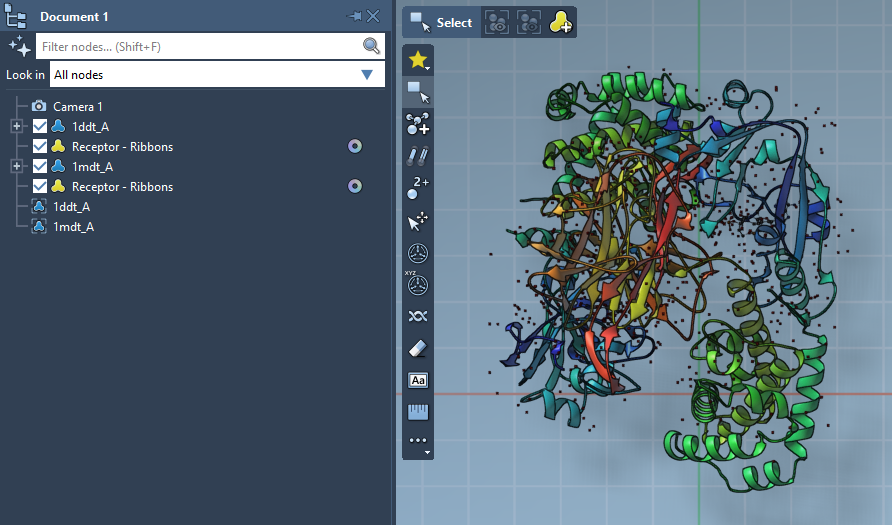
- In the Document view, select your structure (e.g.,
1DDT). - Go to Edit > Conformation and name it (e.g.,
1DDT A).
Repeat for 1MDT, setting its conformation name to 1MDT A. You now have your starting and goal conformations ready for interpolation.
3. Run ARAP Interpolation
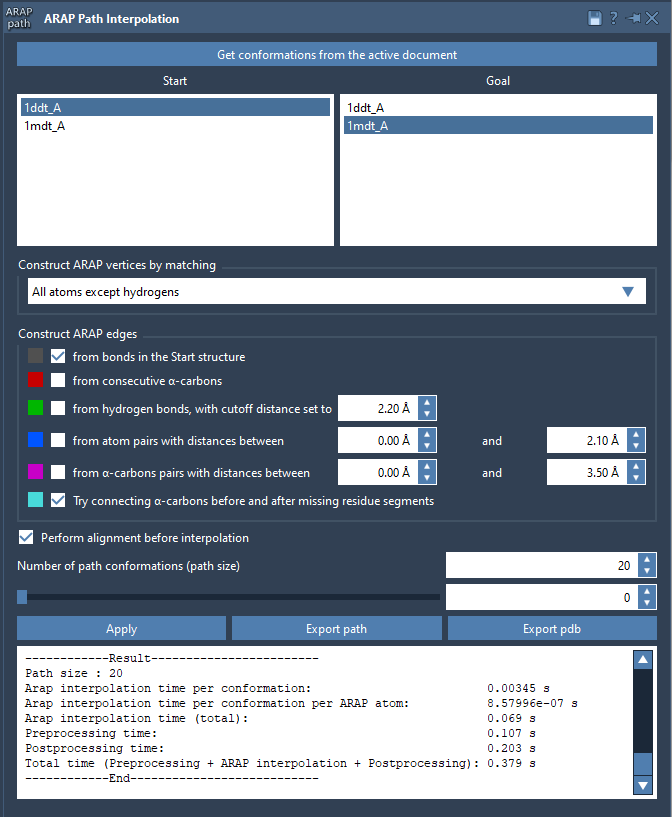
Open the ARAP Interpolation app (Home > Apps > Biology > ARAP Path Interpolation) and configure the following options:
- Start:
1DDT A, Goal:1MDT A. - Matching Atoms: Choose All except hydrogens for atom matching.
- ARAP Edges: Select options such as “from bonds in the Start structure” and “Try connecting α-carbons before and after missing residue segments.”
Ensure alignment by checking Perform alignment before interpolation. Set the “Number of path conformations” to 20, including the start and goal states. This will generate 18 intermediate conformations.
Click Run to compute your transition path, and visualize the stunningly smooth transitions using the built-in slider.
Step 4: Analyze and Export
Once the computation is complete, here’s what you can do:
- Visualize Pathlines: Use the slider in the ARAP app to explore each intermediate conformation.
- Visualize Edge Construction: Toggle visibility of the ARAP edges for deeper insights into the interpolation structure.
- Export Results: Save the full path trajectory as a PDB file for use in simulations or further modeling projects.
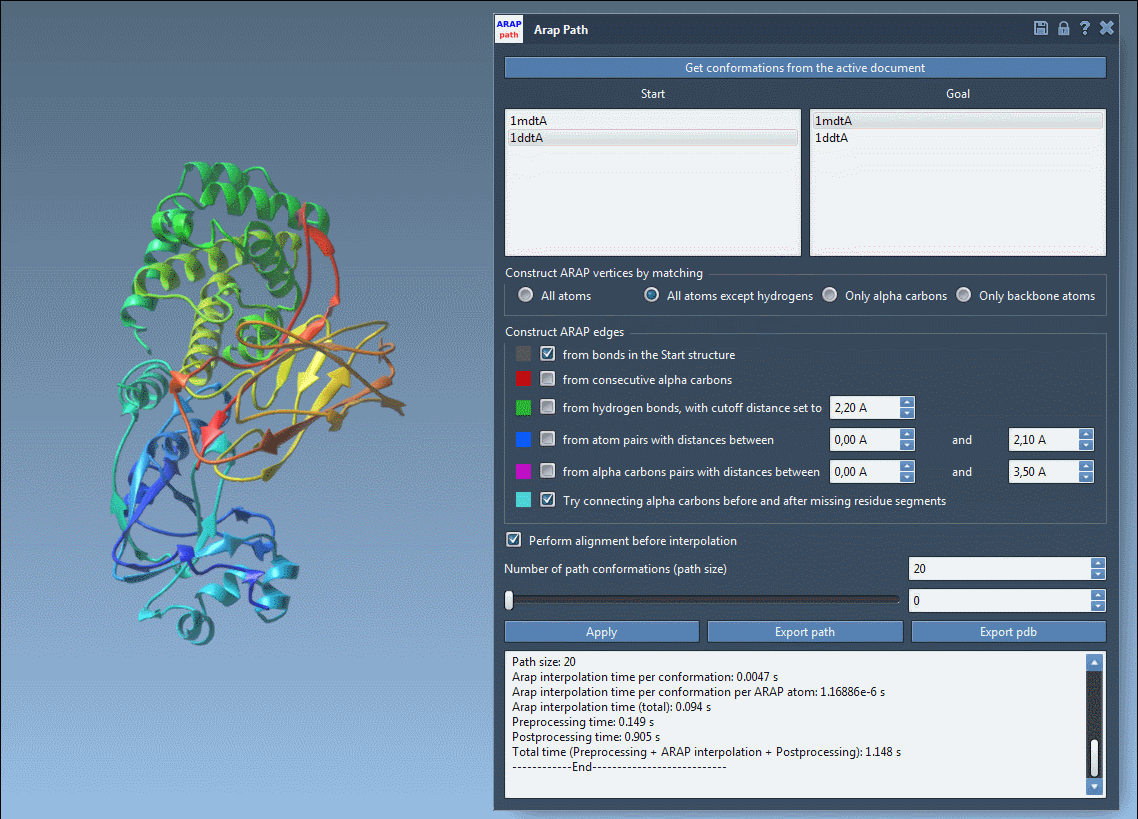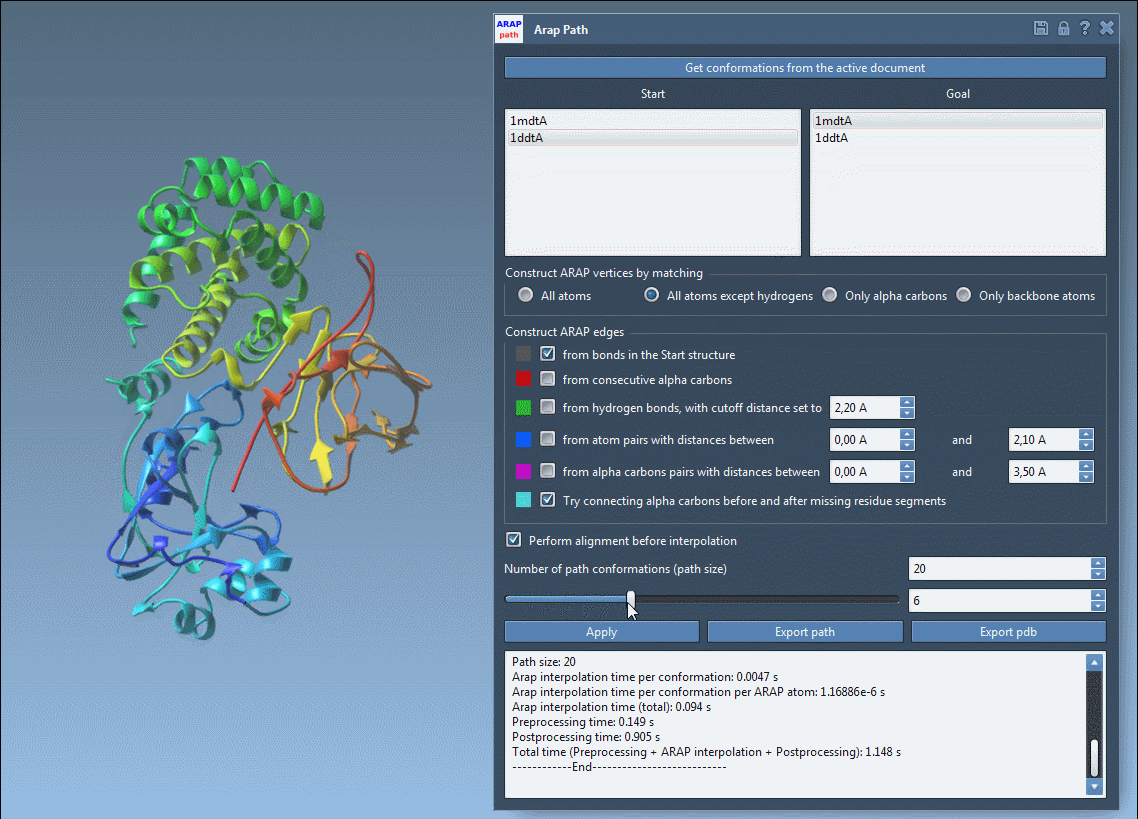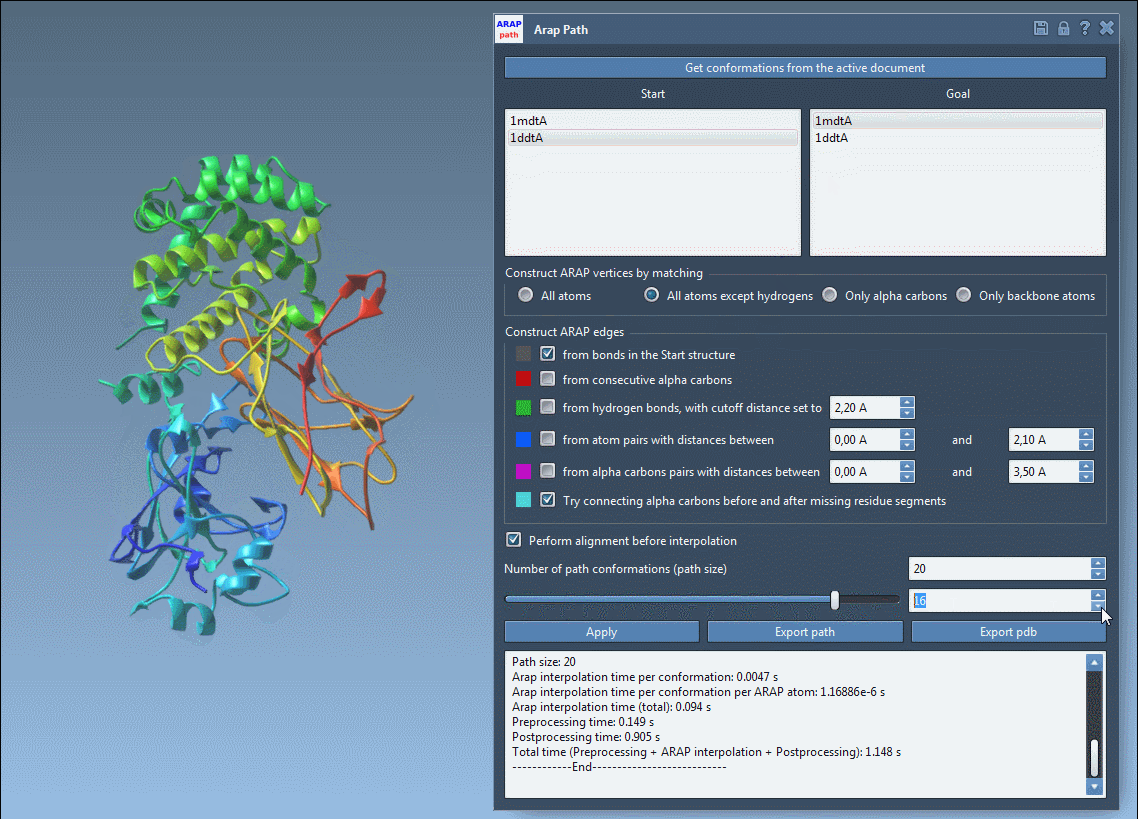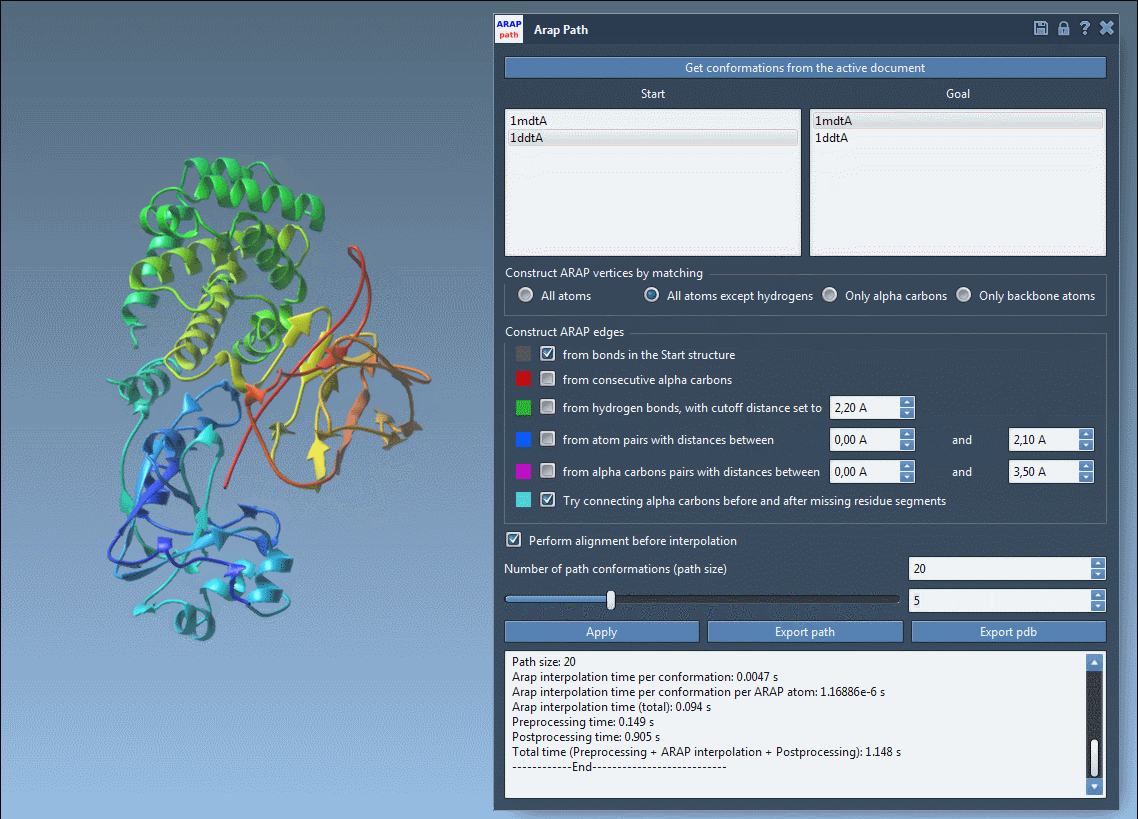
In seconds, you’ve turned your static protein structures into a dynamic, biologically realistic transition path.
Next Steps
The ARAP Interpolation tool isn’t just a standalone feature—it integrates seamlessly with simulation workflows like umbrella sampling (via the GROMACS Wizard) and steered molecular dynamics. By generating informed, intermediate states, ARAP can enhance your precision at every step.
For further details and advanced usage insights, visit the ARAP Interpolation documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at SAMSON Connect.