





One frequent challenge in molecular modeling is understanding how a protein transitions from one structure to another. Traditional molecular dynamics simulations can take a lot of time and computational power—even for visualizing a simple pathway between two known conformations.
If you already have two experimentally resolved or predicted protein structures, how do you generate a smooth, plausible transition path between them without launching a full dynamic simulation? This is where the ARAP Interpolator in SAMSON comes in.
The ARAP Interpolator—short for As-Rigid-As-Possible interpolation—lets you compute continuous, geometrically realistic atomic transitions in a few seconds, creating a smooth morph between two protein conformations. It’s particularly useful for quickly generating reaction coordinates, preparing umbrella sampling setups, or simply visualizing conformational movements.
Who is this for?
This tool is ideal for structural biologists, computational chemists, and students who want quick insight into conformational shifts without running full-scale free energy simulations. If your focus is on understanding motion between crystal structures, comparing model predictions, or exploring hypothetical structural pathways, ARAP interpolation could save you a lot of time.
What do you need?
- Start and end protein structures—e.g., PDB IDs
1DDTand1MDT, representing two conformations of Diphtheria Toxin. - SAMSON installed with the ARAP Interpolator Extension added (just click “Add” on the extension page).
Key ARAP Interpolation Setup Tips
Once you’ve cleaned and prepared your structures, the essential configuration steps in the ARAP Interpolation App are:
1. Choose the conformations
- Use Get conformations from the active document to load the starting and goal conformations.
2. Match Atoms
- Match only non-hydrogen atoms for better performance and fewer artifacts.
- Alternatively, use just α-carbons for a smooth backbone movement.
3. Define ARAP Edges
- Use edges from bonds in the Start structure to preserve standard connectivity.
- Check Try connecting α-carbons before and after missing residue segments to handle missing residues smartly.
4. Align Before Interpolation
- Always enable Perform alignment before interpolation to remove global misalignment between input structures.
5. Generate the Path
- Set the Number of path conformations (e.g., 20 frames).
- Click Run—and you’ll get a smooth interpolation path in seconds.
What does it look like?
You can visualize each interpolated conformation with a slider:
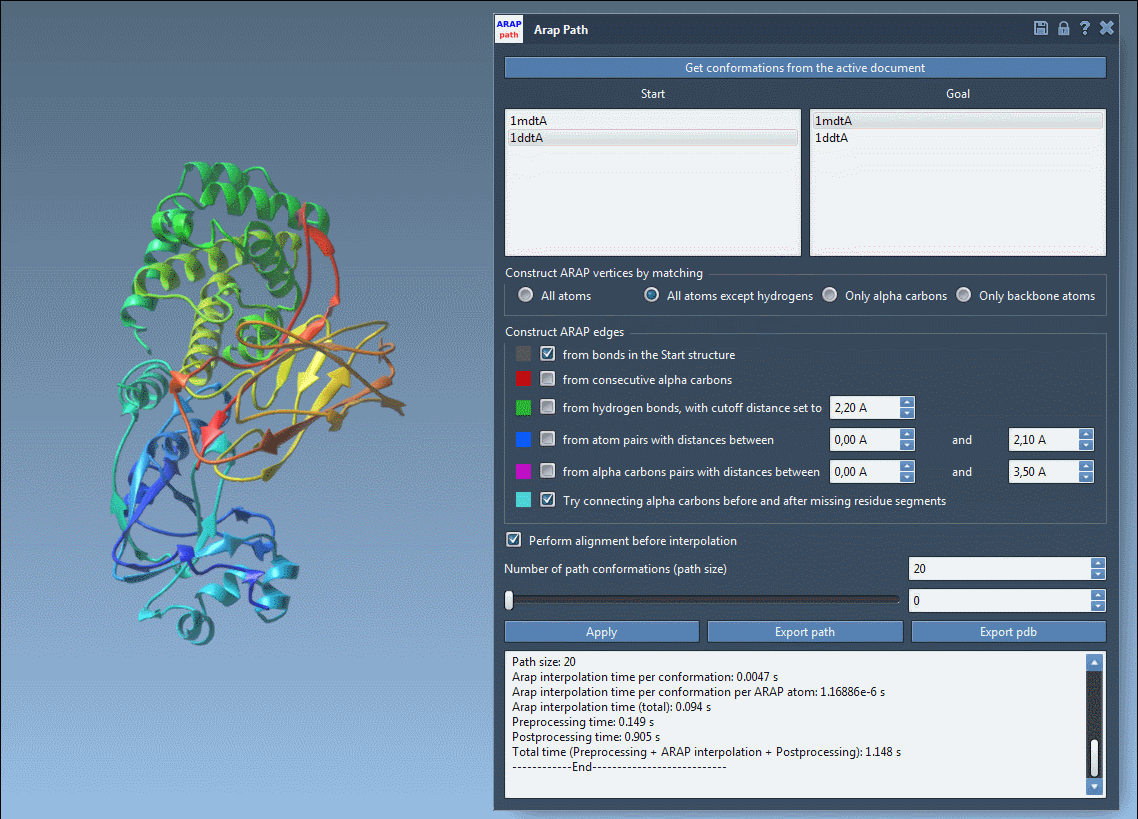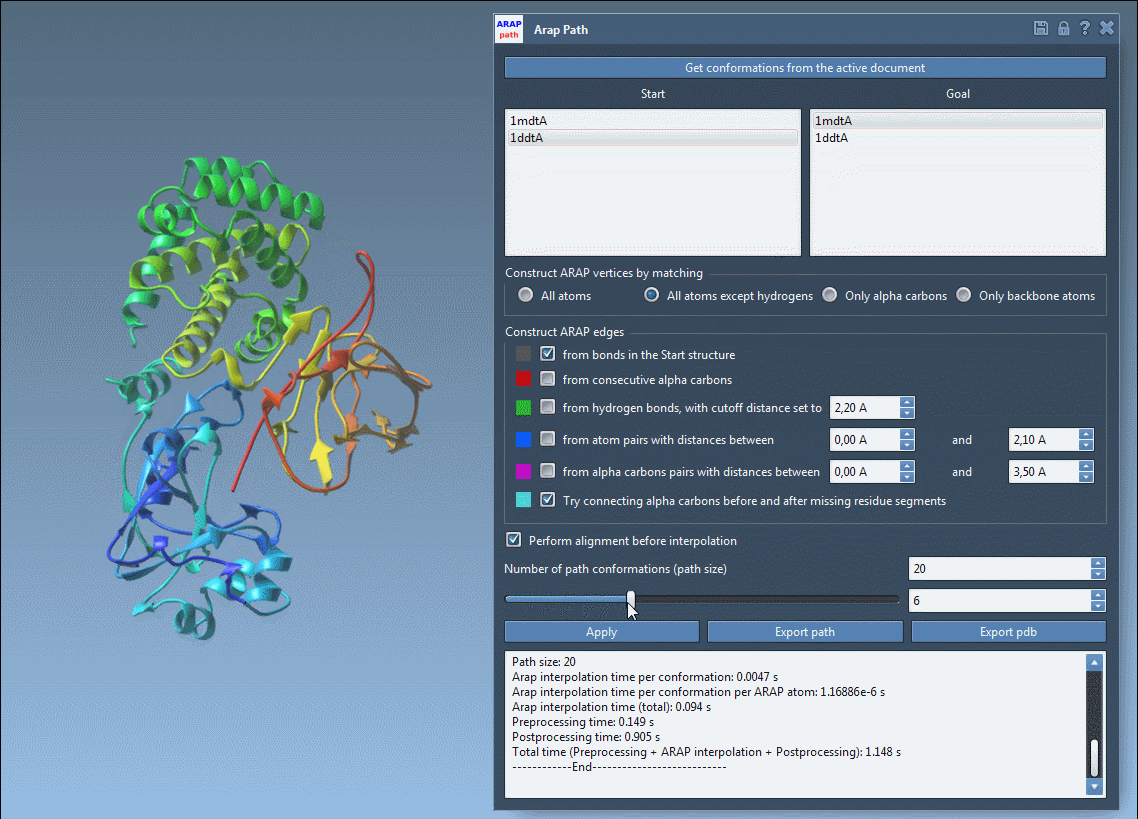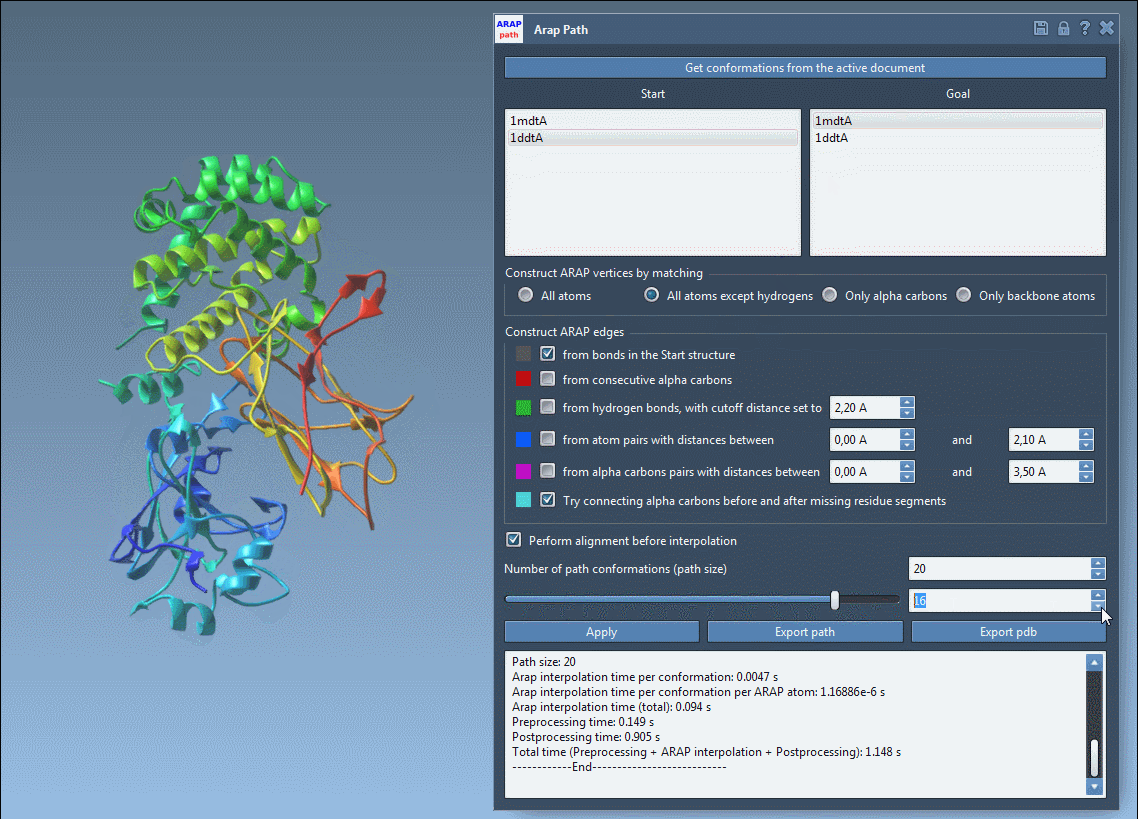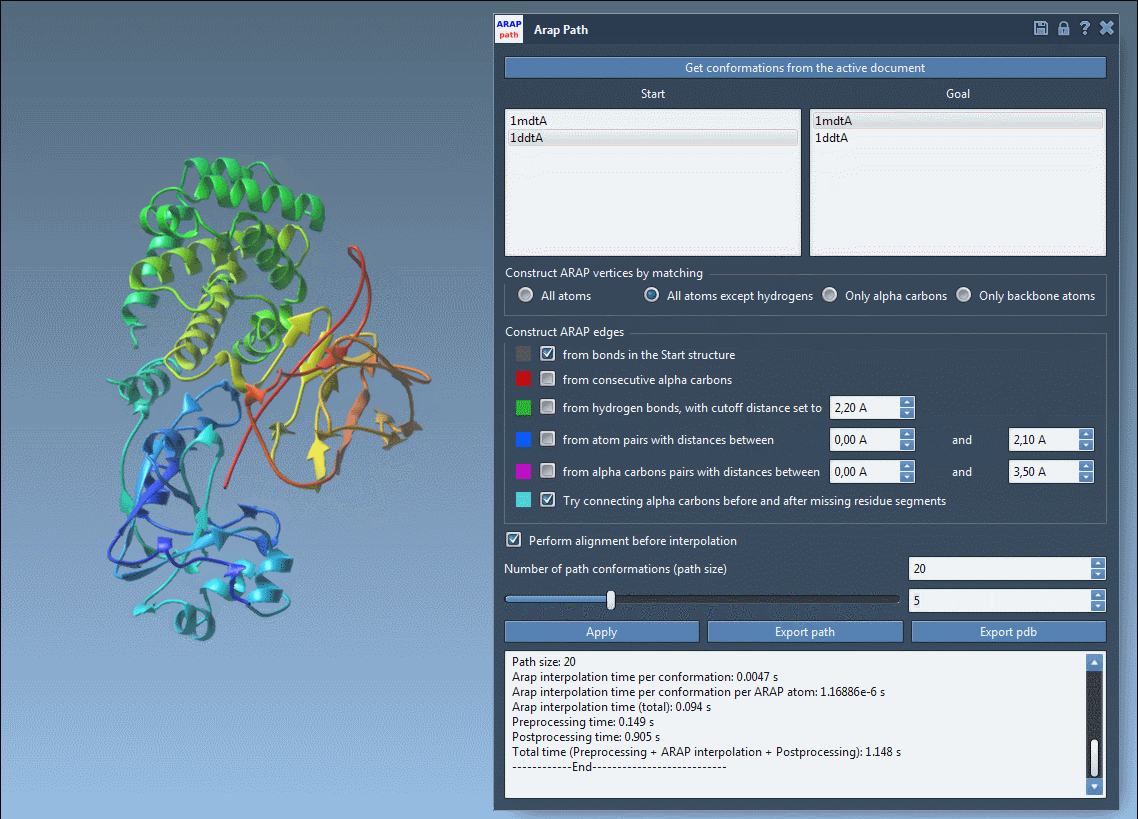
Analyze & Export
You can export the whole trajectory as a PDB file (or as individual conformations). You can also use it as input for simulation workflows like umbrella sampling in the GROMACS Wizard, or further refine it using tools like P-NEB.
Want to Try It?
To explore the full tutorial and get detailed visuals of each step, visit the documentation: ARAP Interpolation for Protein Structures.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON at https://www.samson-connect.net.