





Setting up molecular simulations can be a challenging task, but SAMSON’s Universal Force Field (UFF) interaction model introduces an efficient and user-friendly way to bridge complex molecular mechanics and intuitive modeling. If you’ve ever struggled with setting bonds, atom types, and interaction models, this guide explores SAMSON’s UFF setup process to ease your workflow.
The Problem: Streamlining Molecular Simulations
Molecular modelers often face hurdles in preparing simulation-ready molecular systems. Tasks like defining covalent bonds, bond orders, and atom types manually can be highly time-consuming and error-prone. SAMSON addresses this by integrating the Universal Force Field—automating these critical steps while offering configurability to advanced users.
Step-by-Step UFF Setup
Here’s how to quickly get up and running with UFF in SAMSON:
- Open your molecular system: Start by selecting the system you want to simulate. Ensure it’s loaded into SAMSON.
- Add the UFF simulator: Go to
Edit > Simulate > Add simulator, or use the shortcutCtrl+Shift+M(Windows) /Cmd+Shift+M(Mac). Select ‘Universal Force Field’ from the list of interaction models. - Select a state updater: Choose a state updater like the Fast Inertial Relaxation Engine (FIRE) to optimize geometry during simulations.
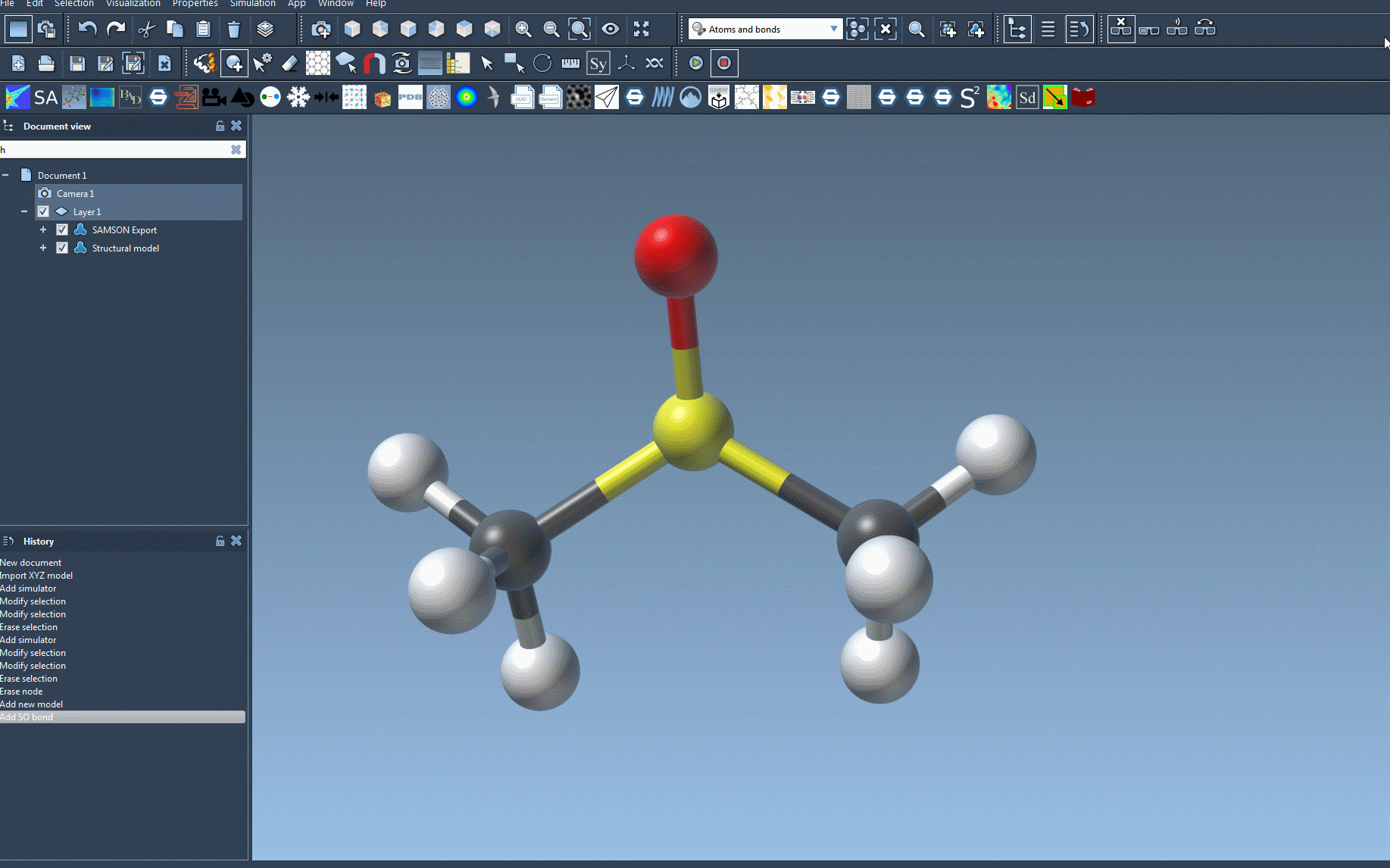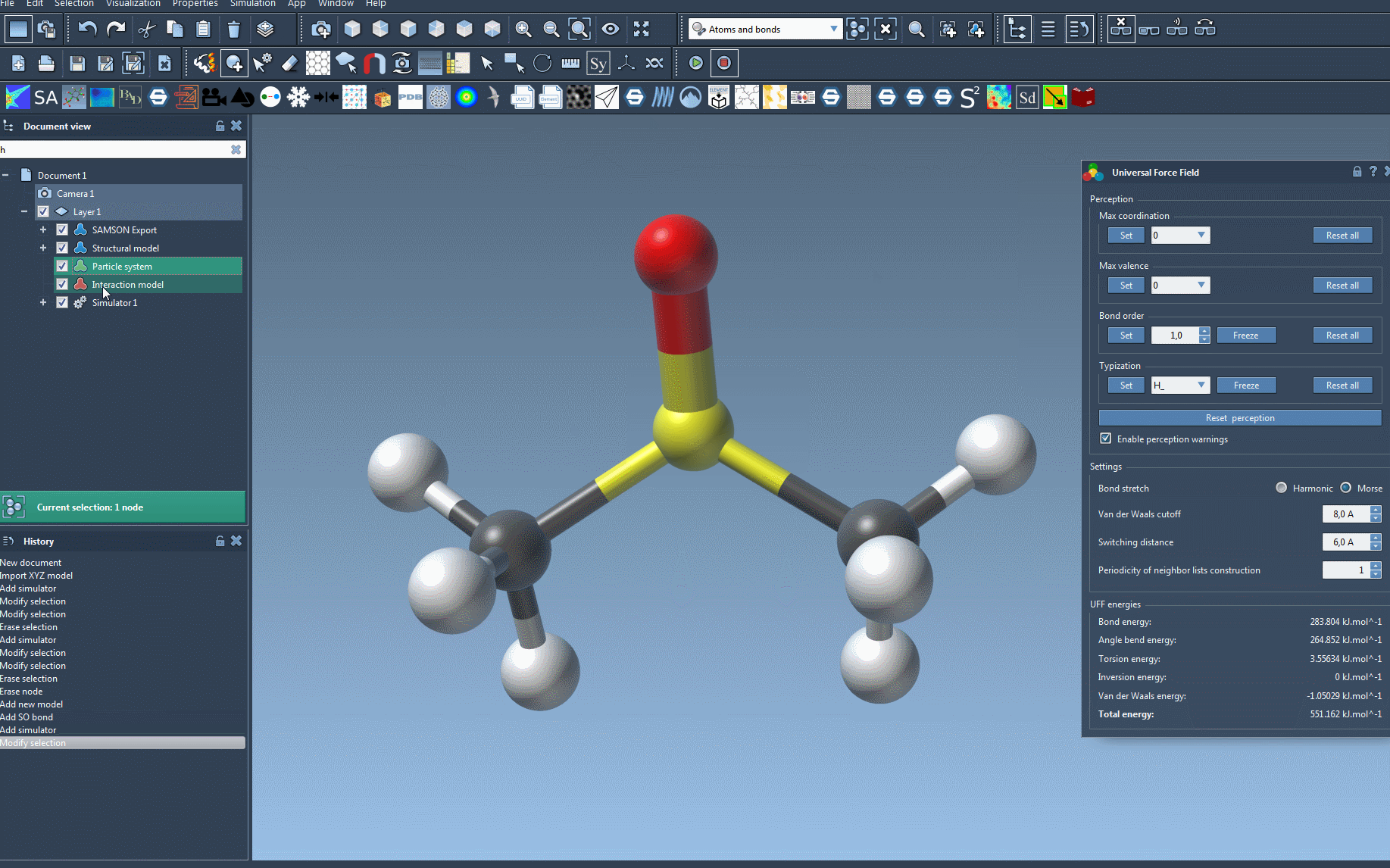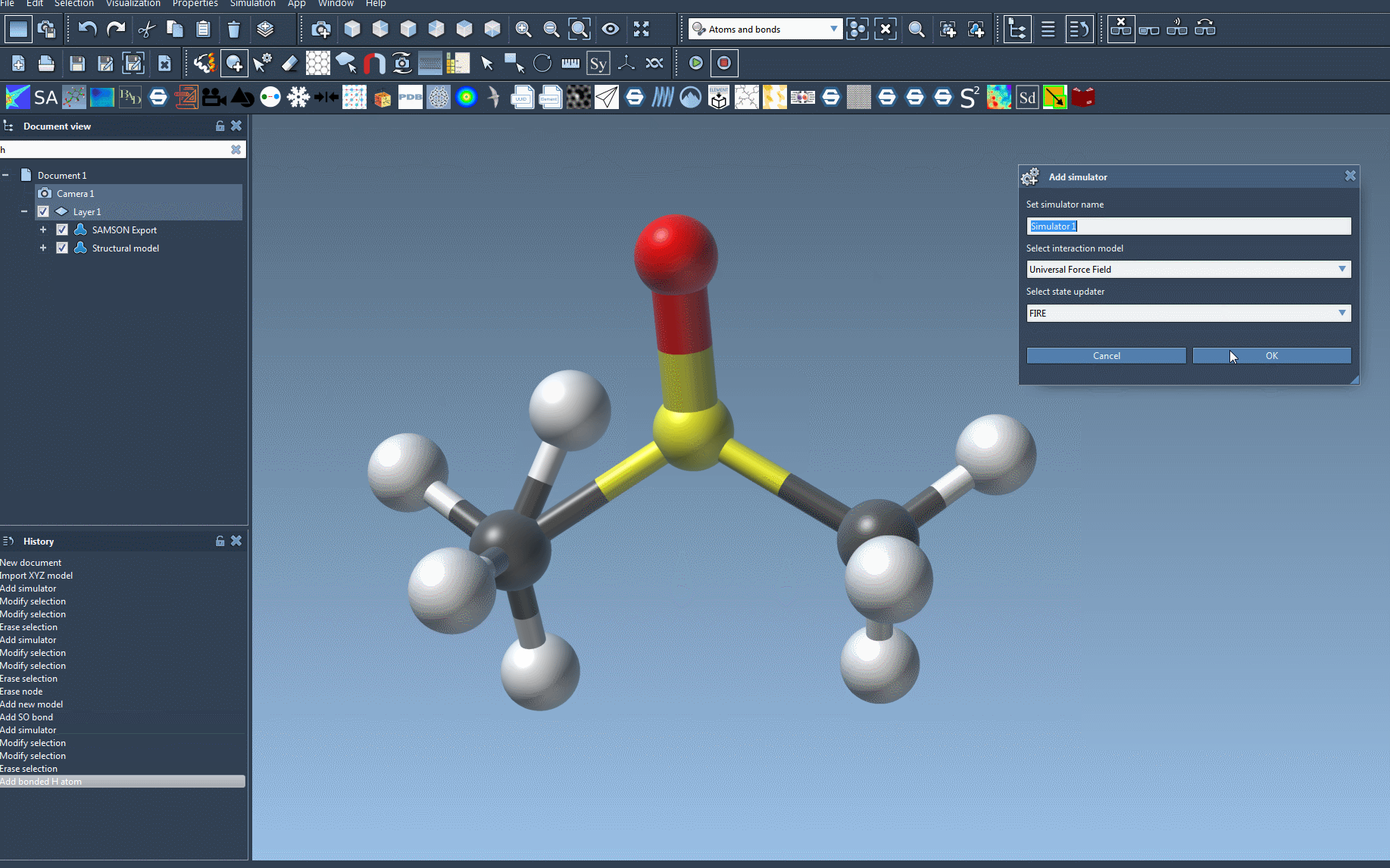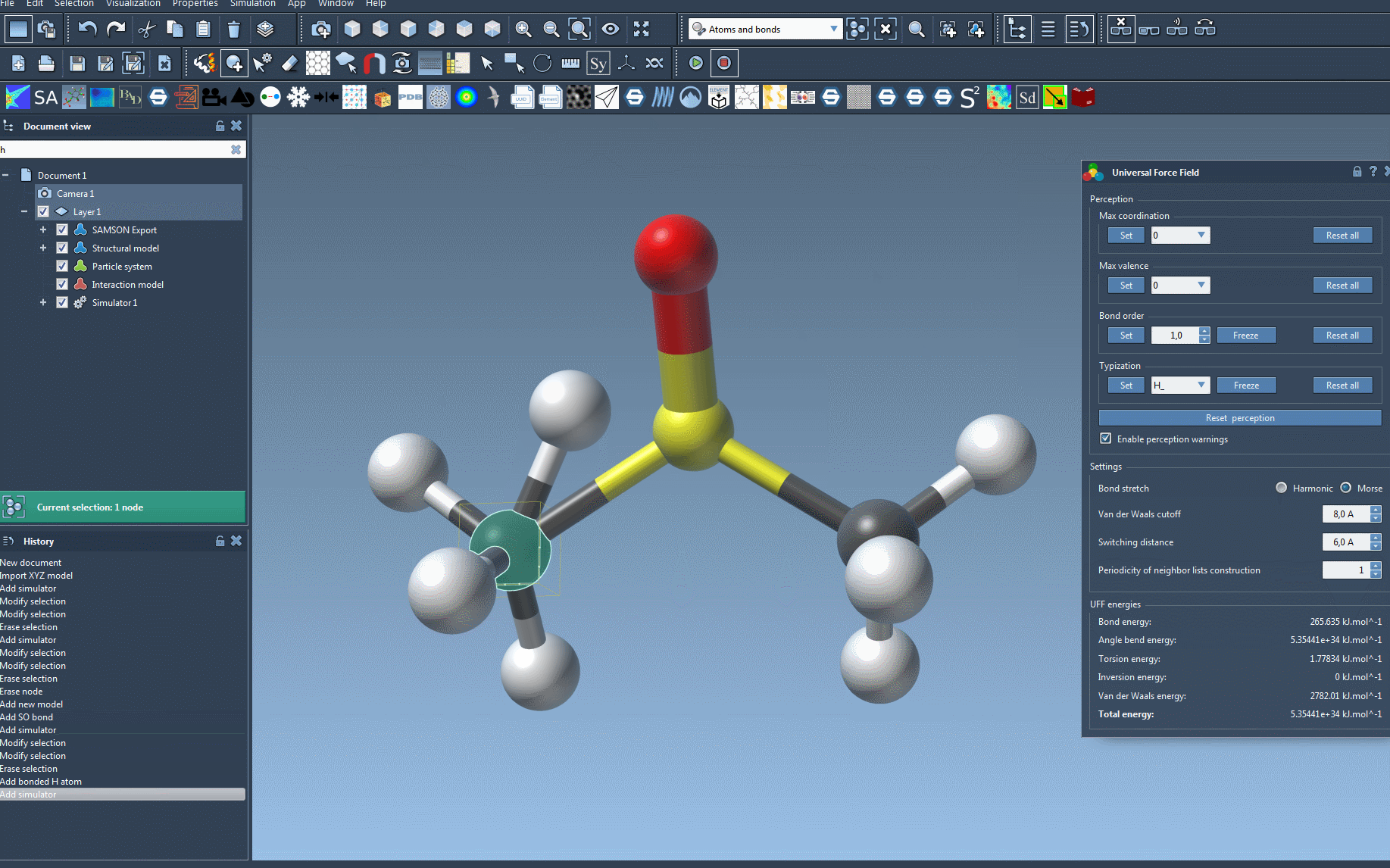
- Configure bonds: In the UFF setup window, you can choose to use existing bonds in the system or allow UFF to recompute them based on atom positions and types.
Once set up, the module automatically handles the computational aspects like covalent bond generation, bond order calculation, and atom type determination. This automation helps avoid inconsistencies while offering faster simulation readiness.
Customization Options for Advanced Users
While the default settings are powerful enough for most tasks, SAMSON also supports advanced customization:
- Bond Stretch Interaction: Toggle between ‘Harmonic’ and ‘Morse’ for bond stretch interaction to fit your simulation needs.
- Van der Waals (vdW) Cutoff: Adjust the cutoff distance for vdW interactions. You can also fine-tune the switching distance to smoothly weight calculations beyond a certain range.
- Neighbor List Periodicity: Modify the frequency of neighbor list recalculations in vdW interactions for performance optimization.
An intuitive interface ensures you can monitor how the system reacts, showing changes in energy components in real-time. You can even interactively move atoms to see the immediate impact on the system.
Efficient Error Handling
What if inconsistencies are detected? If UFF identifies mismatched bonds or atom types during setup, it will notify you with a detailed warning. This proactive approach minimizes potential simulation errors, allowing you to address issues before proceeding.
Why Use SAMSON for UFF Simulations?
SAMSON’s UFF integration saves significant time for modelers by automating tedious molecular structure tasks while providing flexibility for fine-tuning. With a few simple clicks, you can set up and execute simulations, visualize energy changes, and achieve robust molecular modeling with minimal effort.
Learn more about UFF in SAMSON by visiting the official documentation.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON at SAMSON Connect.