





Understanding molecular motion is crucial in structural biology and molecular design, especially when studying protein-ligand interactions or conformational transitions. But modeling flexible structures like the SARS-CoV-2 spike protein can be technically challenging and time-consuming. Knowing how to simulate such motions intuitively and visually is a frequent pain point for many molecular modelers.
Luckily, an insightful post from the SAMSON documentation introduces a practical way to visualize the conformational transition of the SARS-CoV-2 spike protein—specifically, its motion from the closed (inactive) to the open (receptor-binding) state. This motion is key for understanding how the virus recognizes human cells.
The Closed → Open Transition
The SARS-CoV-2 spike protein enables the virus to enter human cells by binding to the ACE2 receptor. But it can only do so in its open conformation. Therefore, modeling how the spike transitions from closed to open is a meaningful and informative step in understanding this mechanism at the atomic level.
The visualizations created with the SAMSON platform provide clear, dynamic views of how this motion unfolds. Using known structural data (PDB entries 6VXX for the closed state and 6VYB for the open state), intermediate conformations were generated using interpolation and minimization tools available in SAMSON.
Clear Dynamic Views
Here are several GIFs from SAMSON’s animations showing the spike transition from different angles:
- Side view:
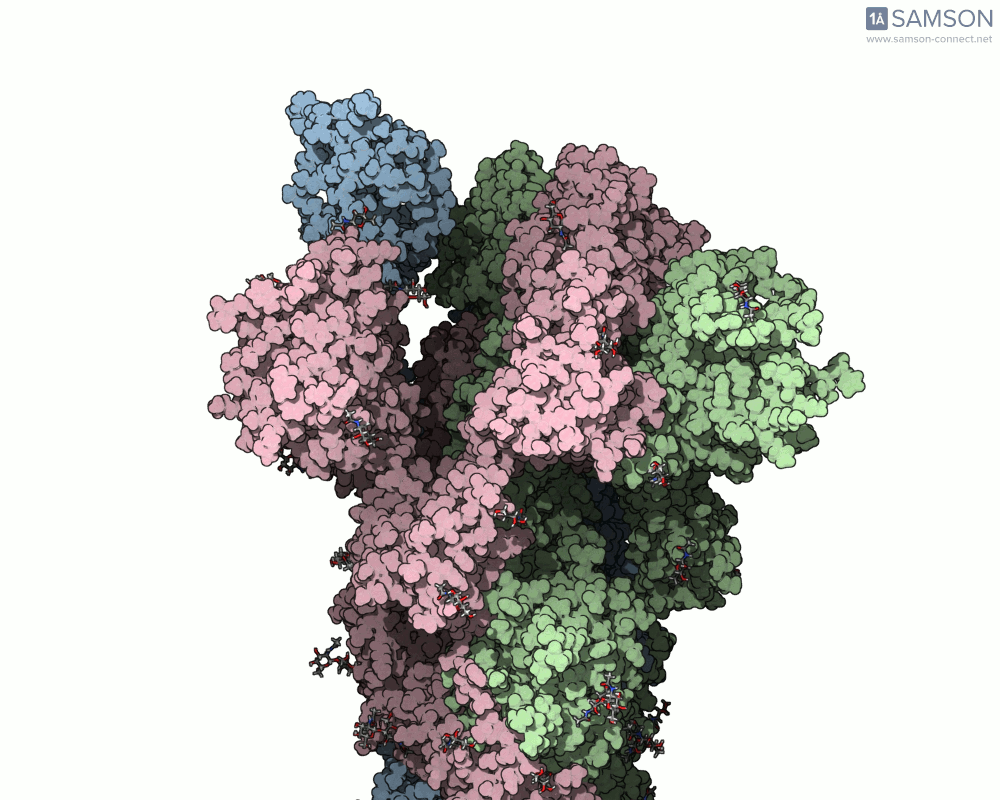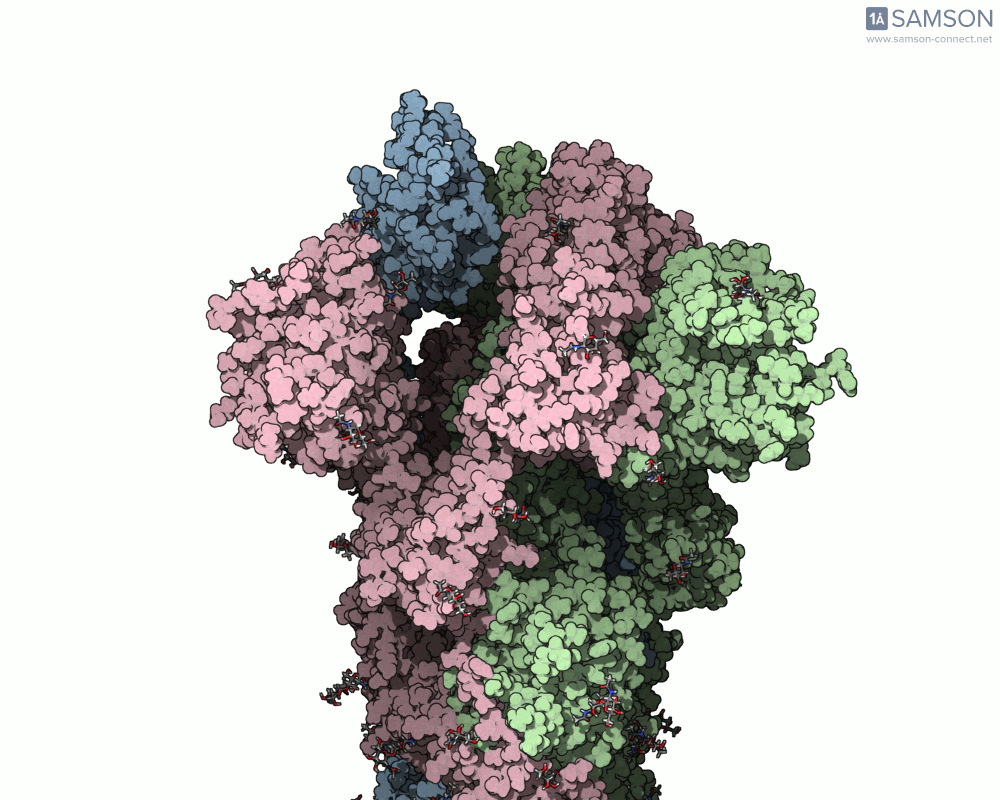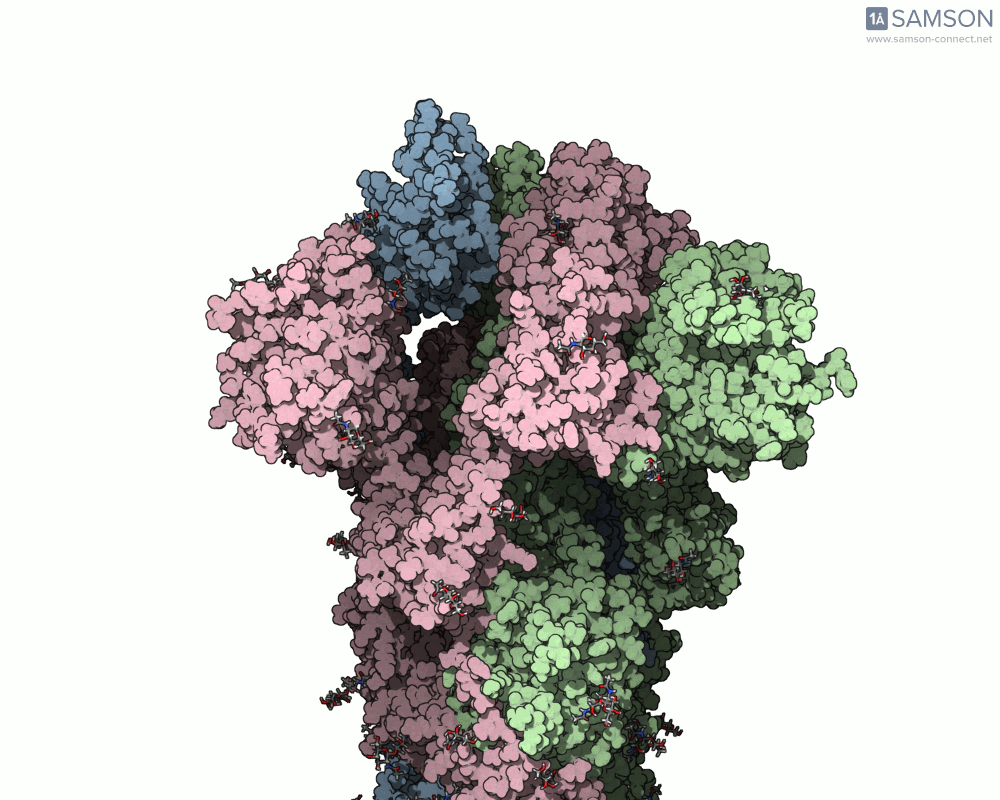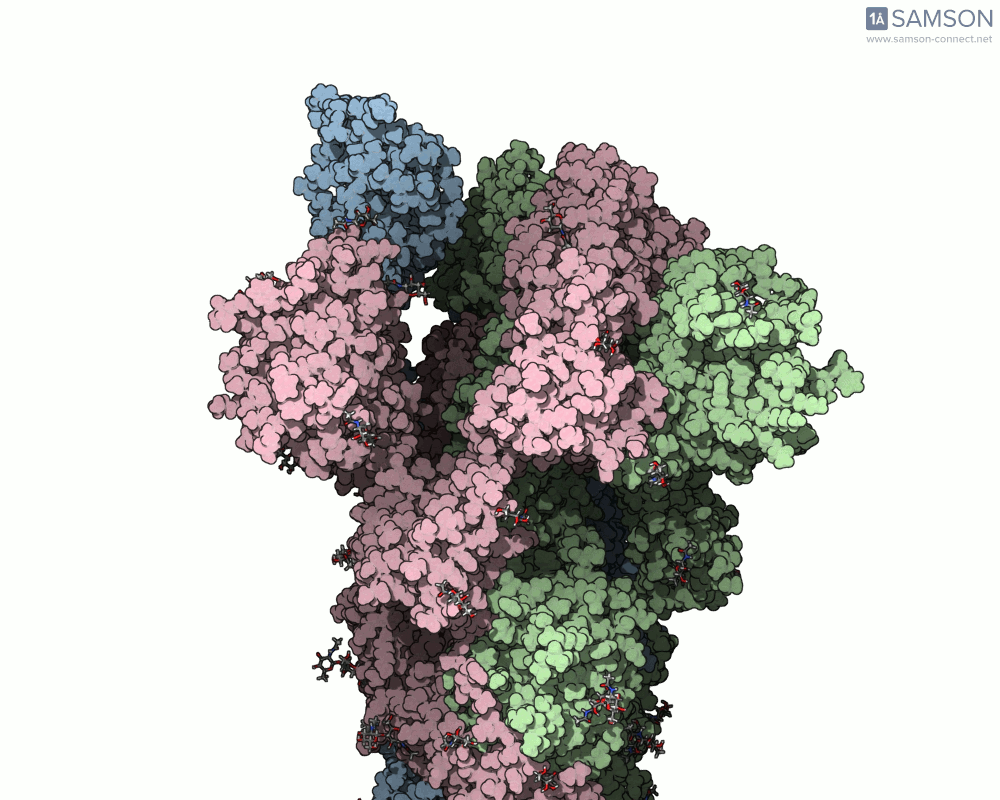
- Oblique angle view:
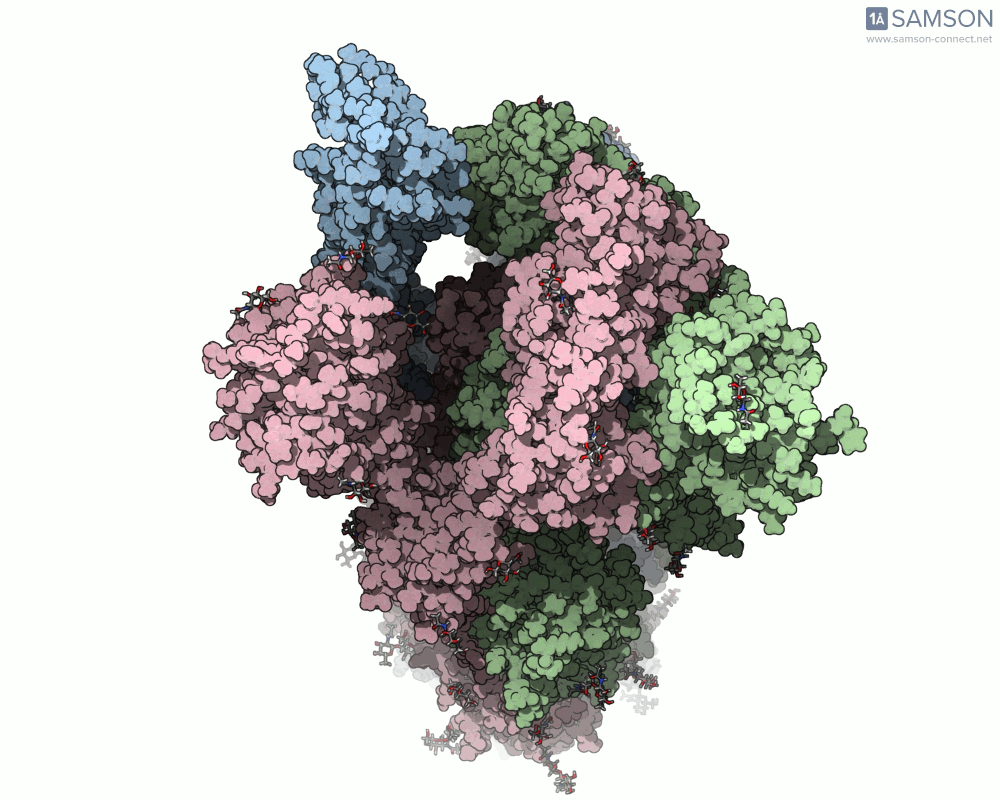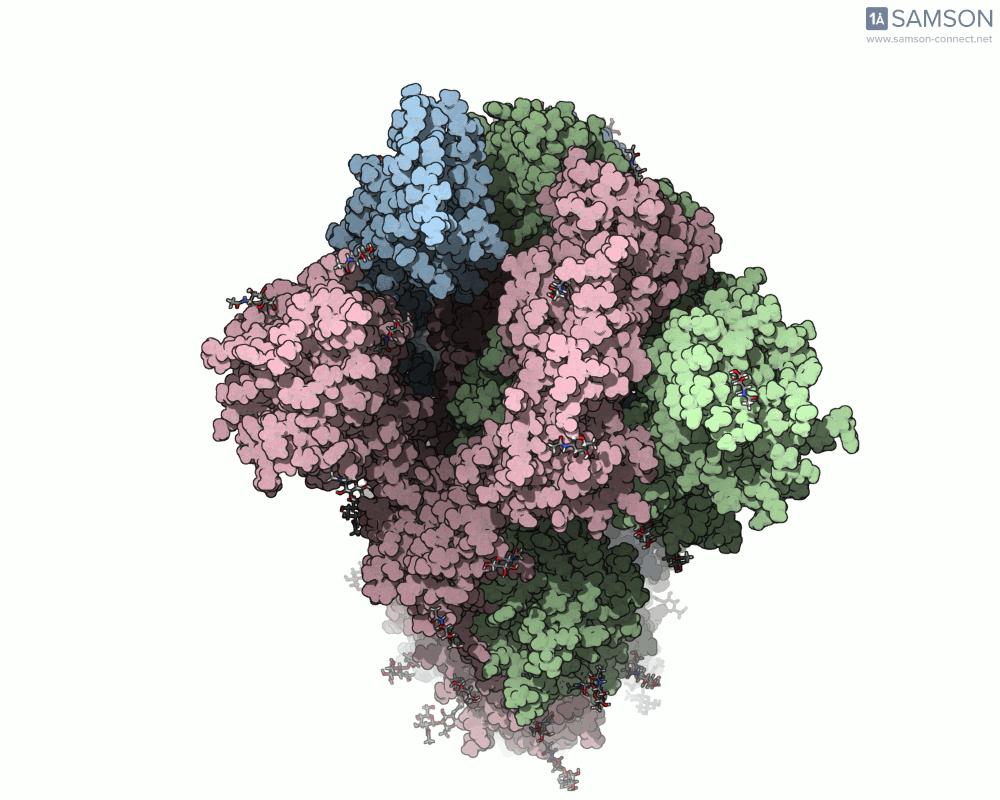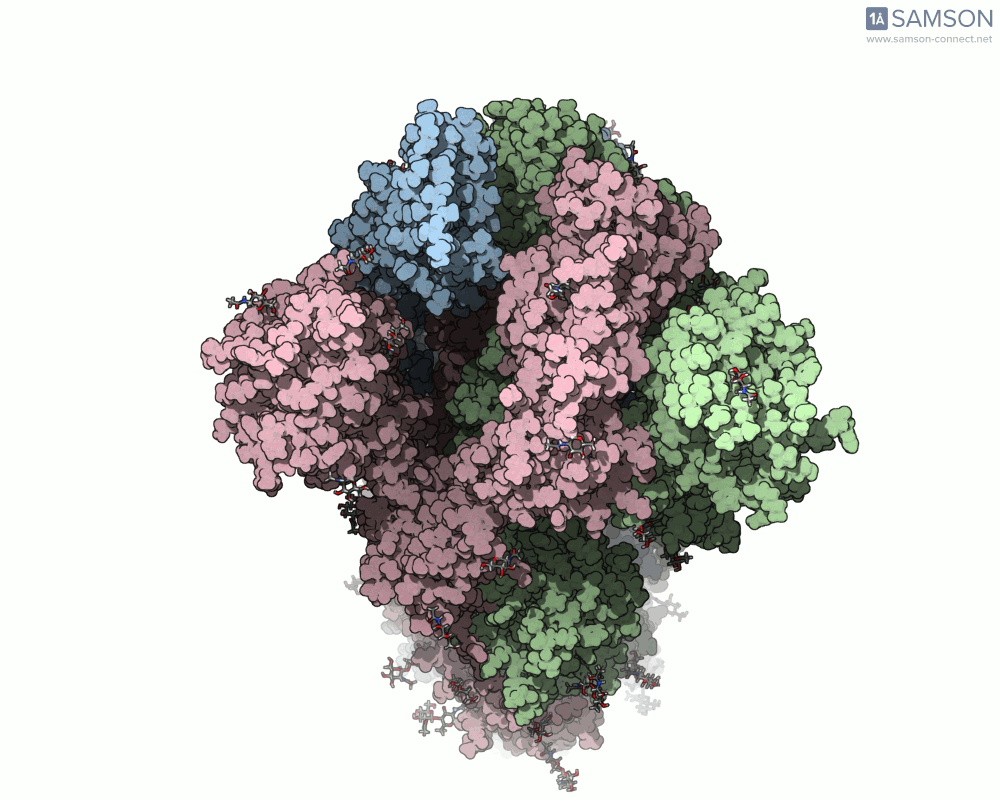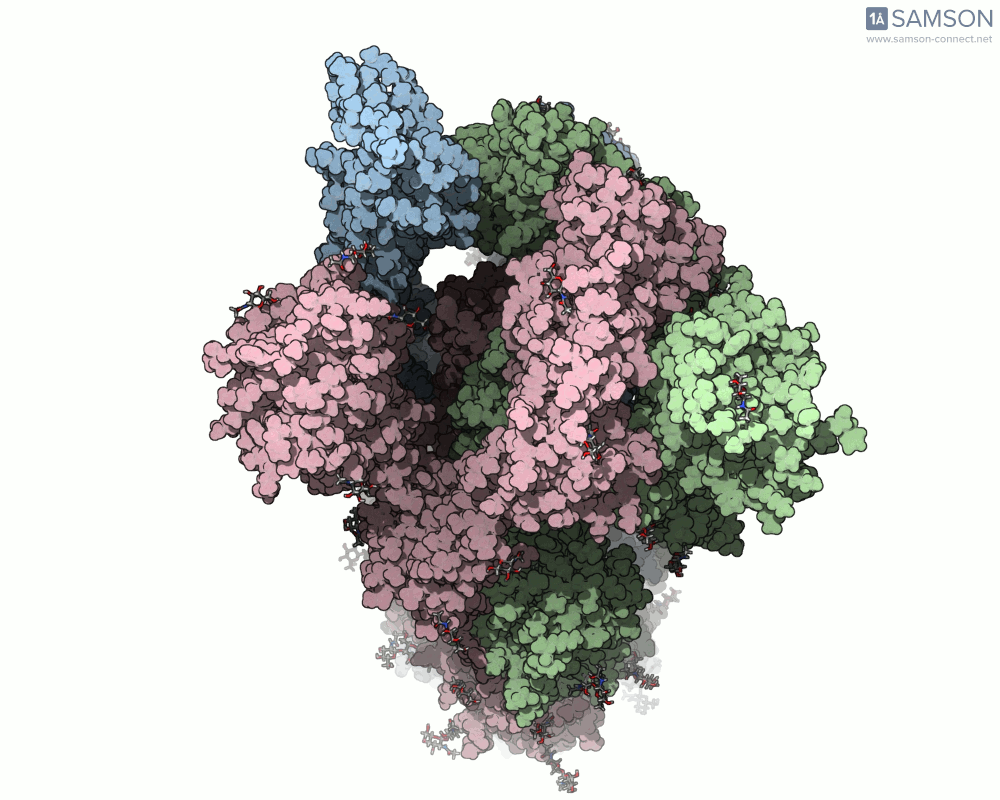
- Top view:
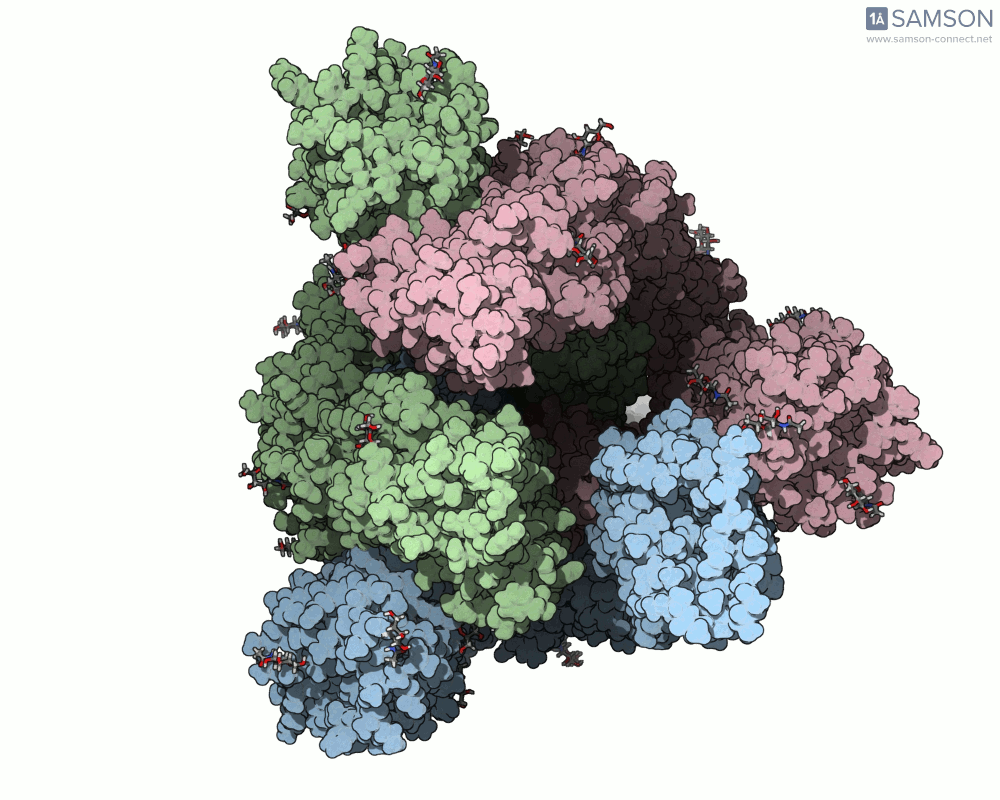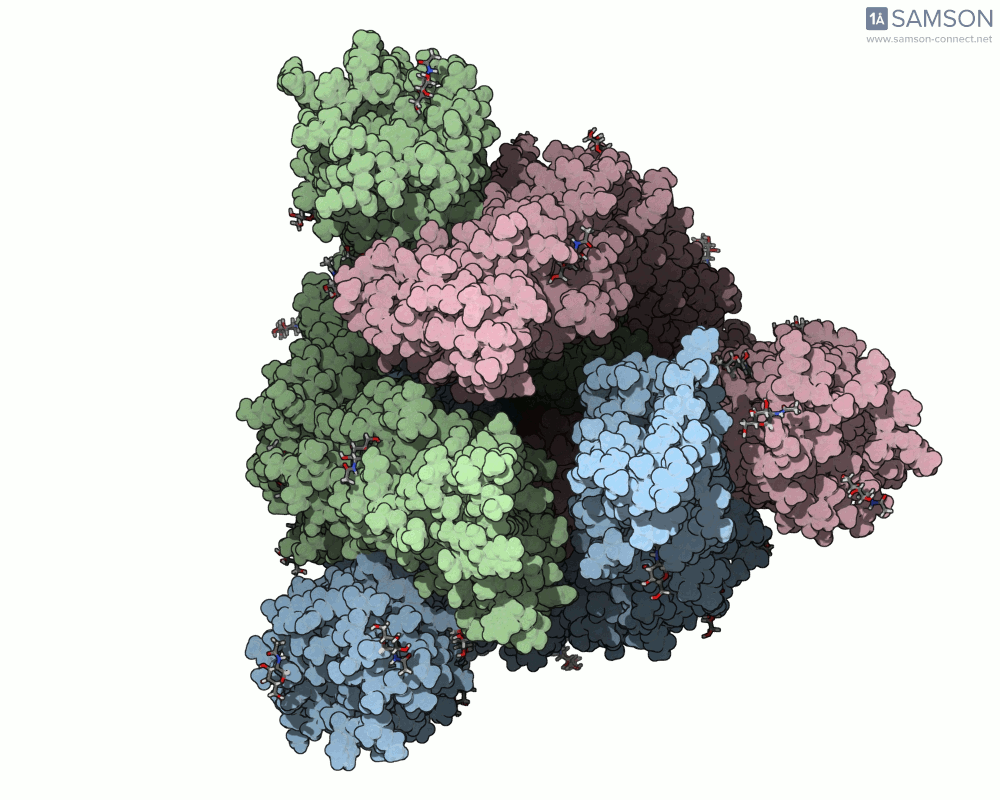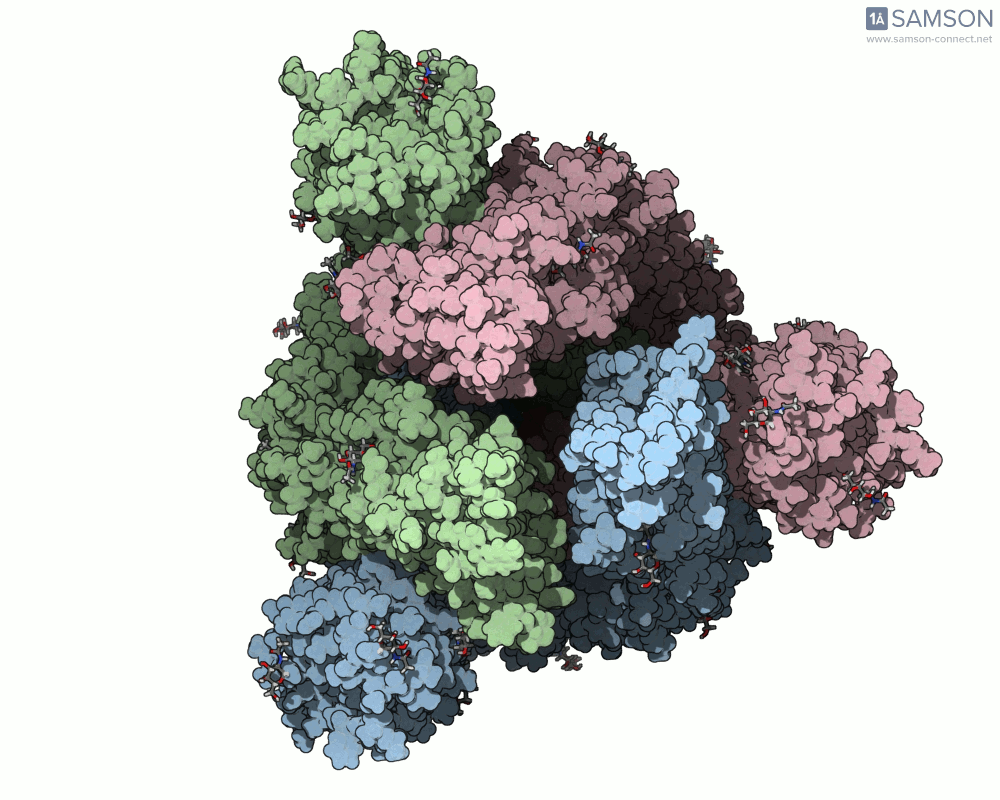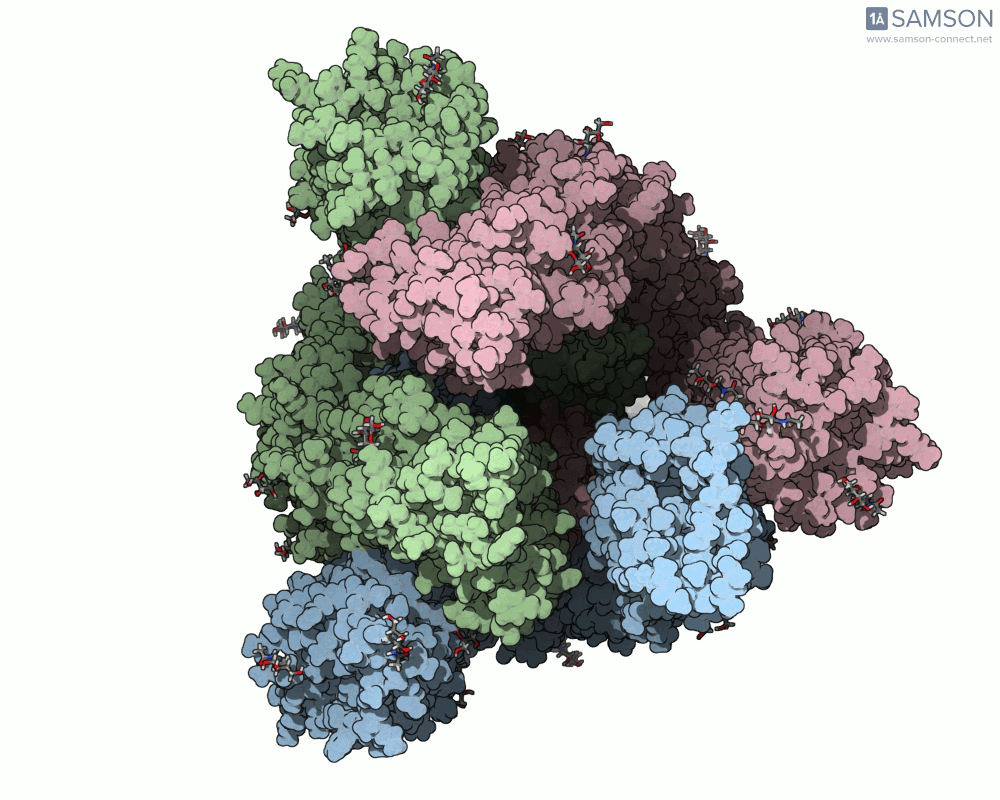
Why This Matters
For modelers working on drug discovery or antibody design, visualizing these transitions is more than just illustrative—it provides contextual information for locating binding sites and predicting structural flexibility that may affect molecular interactions.
Moreover, understanding how the spike exposes the receptor-binding domain helps researchers identify accessible regions for targeting with small molecules or biologics.
How It Was Computed
To compute this transition, SAMSON used a combination of ARAP (As-Rigid-As-Possible interpolation) and P-NEB (Parallel Nudged Elastic Band) algorithms. Here’s a simplified version of the pipeline:
- Prepare the open and closed spike structures (add hydrogens, adjust glycan chemistry).
- Use ARAP interpolation to generate an initial motion path.
- Post-process the path with P-NEB to smooth and refine the motion.
The best part? This pipeline ran in under 20 minutes on a regular laptop. Users can download the trajectory in multiple formats (PDB and SAM), making it accessible for further visualization and processing in other platforms as well.
These visualizations are available under a CC BY 4.0 license, making them freely reusable for research and education. Whether you’re working on viral dynamics, vaccine design, or receptor interaction modeling, having access to resources like these can accelerate your workflows and improve collaboration across teams.
Want to see it in motion and experiment with the files yourself? Check out the full documentation, including downloadable trajectories and instructions, here: SAMSON Documentation.
SAMSON and all SAMSON Extensions are free for non-commercial use. If you’d like to experiment with these tools yourself, you can download SAMSON from https://www.samson-connect.net.