





When designing new drug candidates or optimizing a lead compound, molecular modelers often face the challenge of evaluating how small chemical modifications affect binding affinity, selectivity, or physicochemical properties. One effective approach to address this is positional analogue scanning, where small changes—such as replacing a hydrogen with a nitrogen or adding a fluorine atom—are introduced systematically across the molecule.
This process can be time-consuming and error-prone if done manually. But did you know you can streamline it directly in SAMSON, using the SMILES Manager extension?
Start from a single molecule
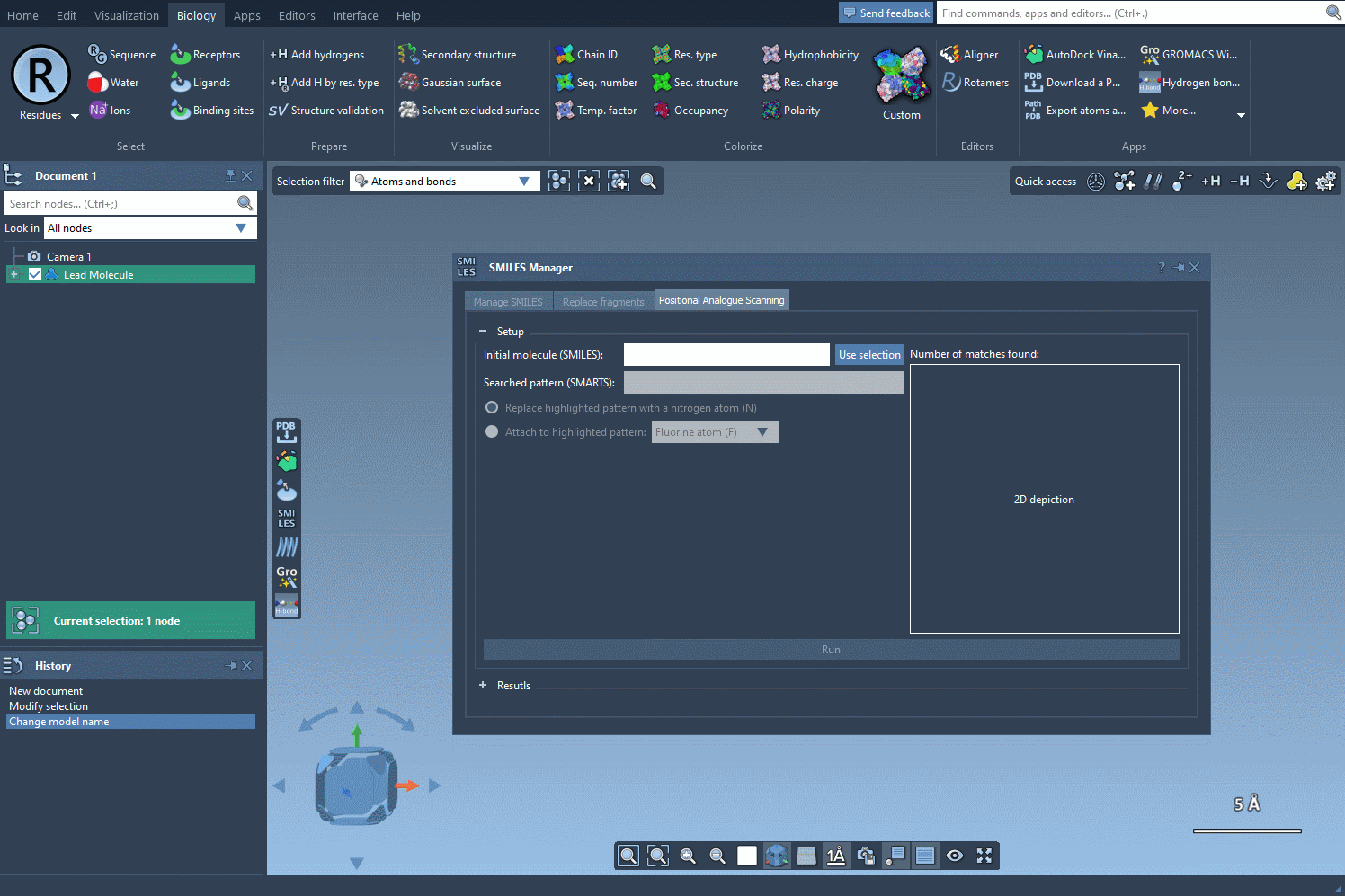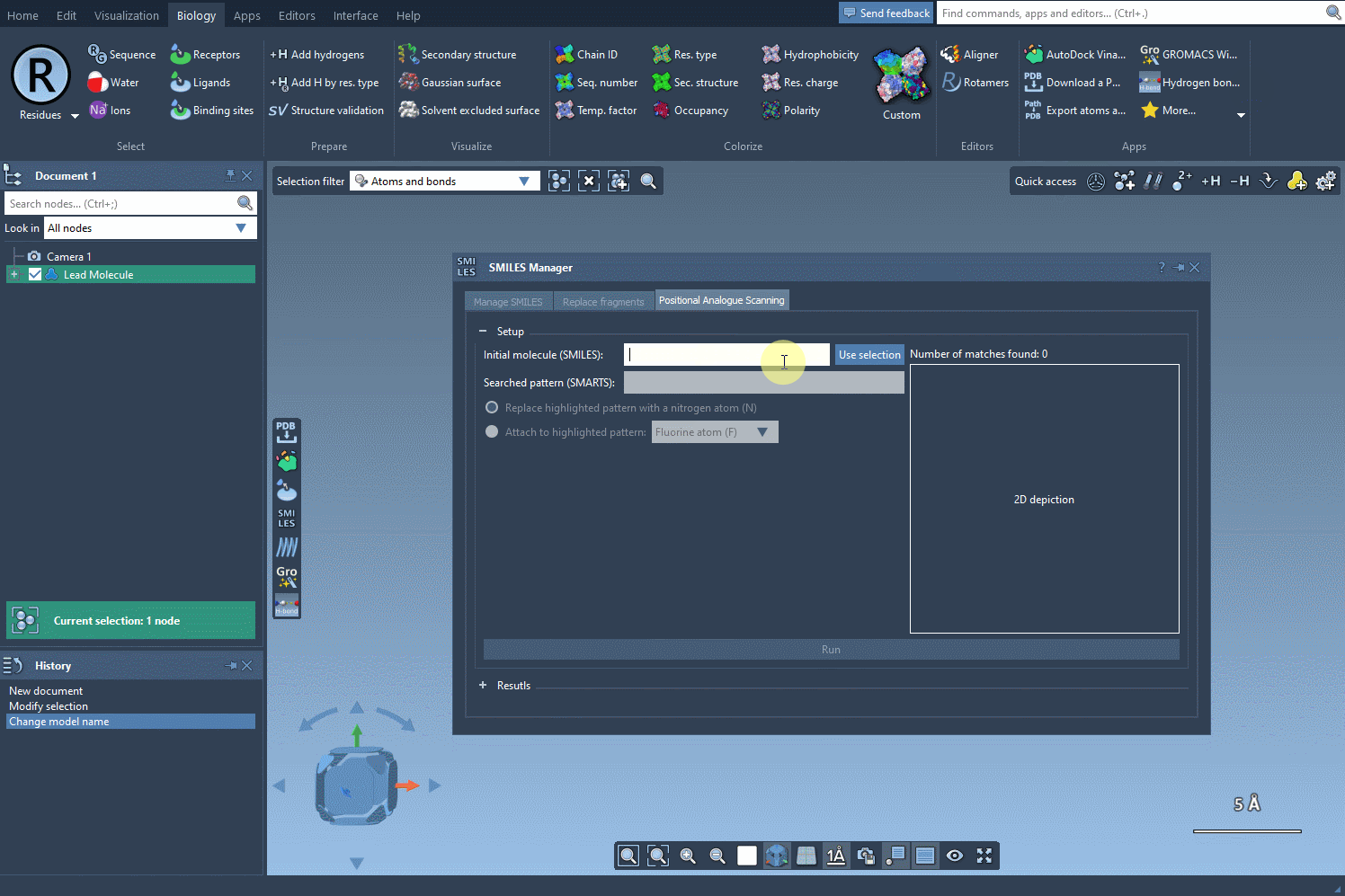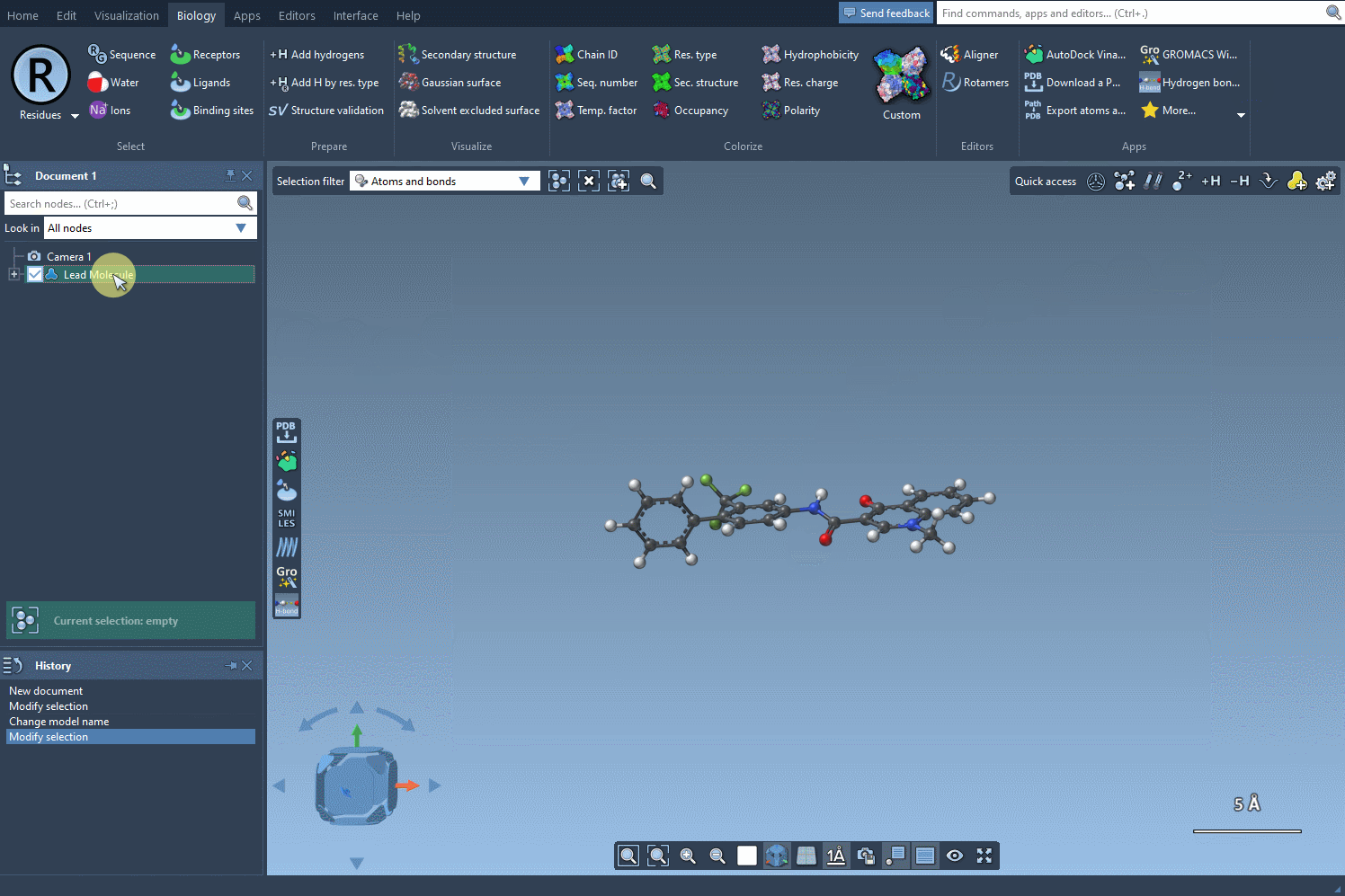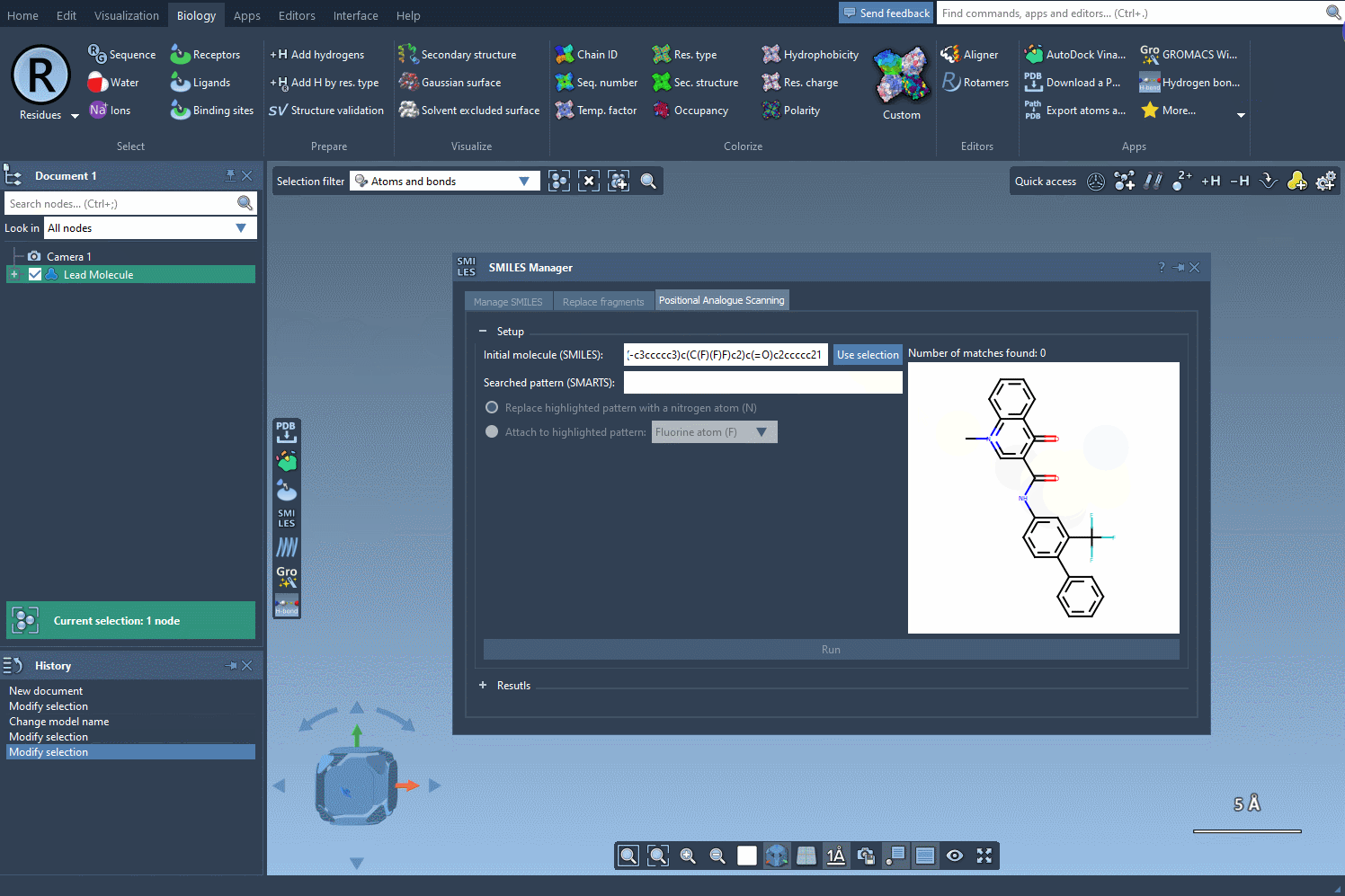
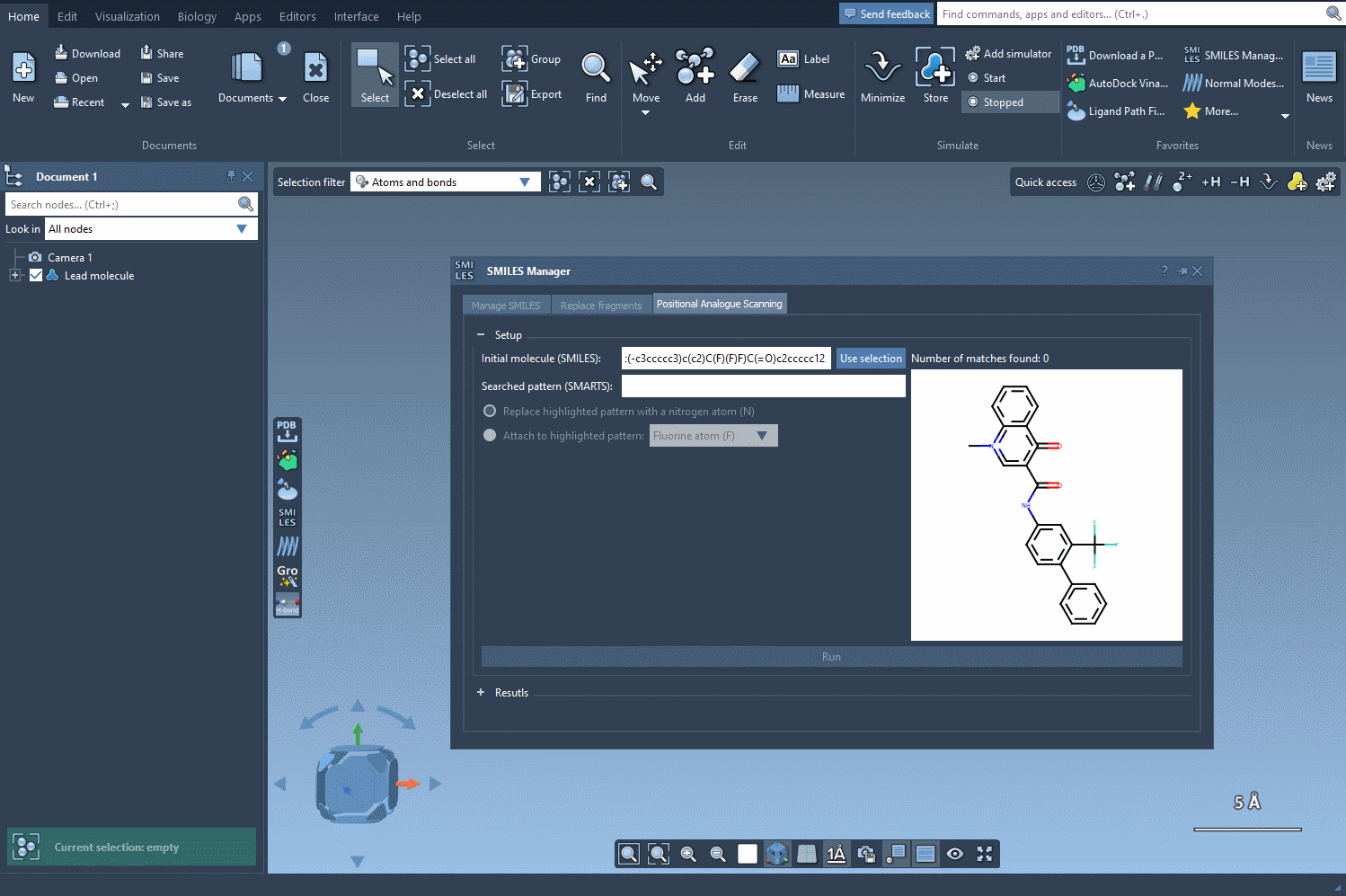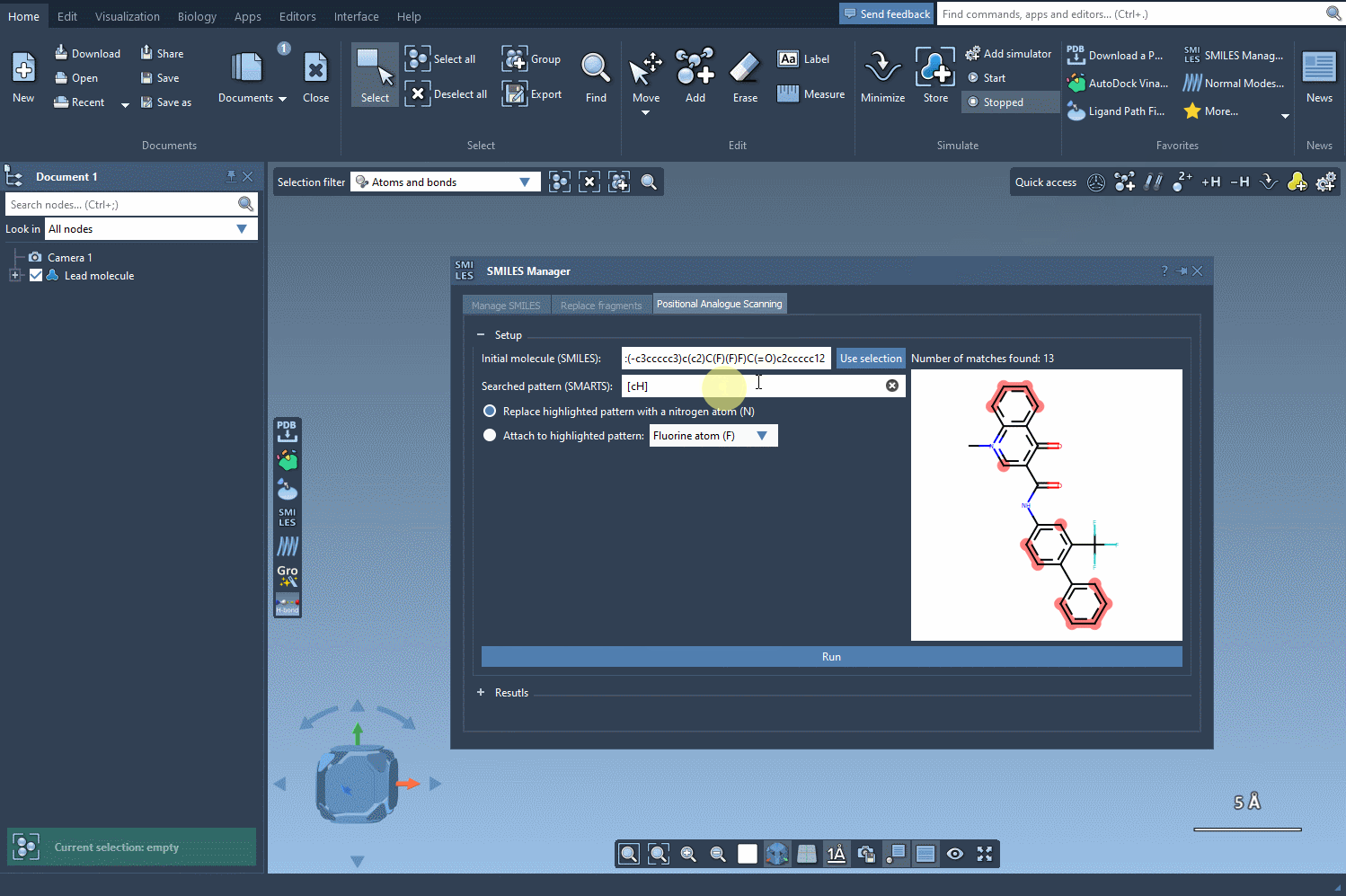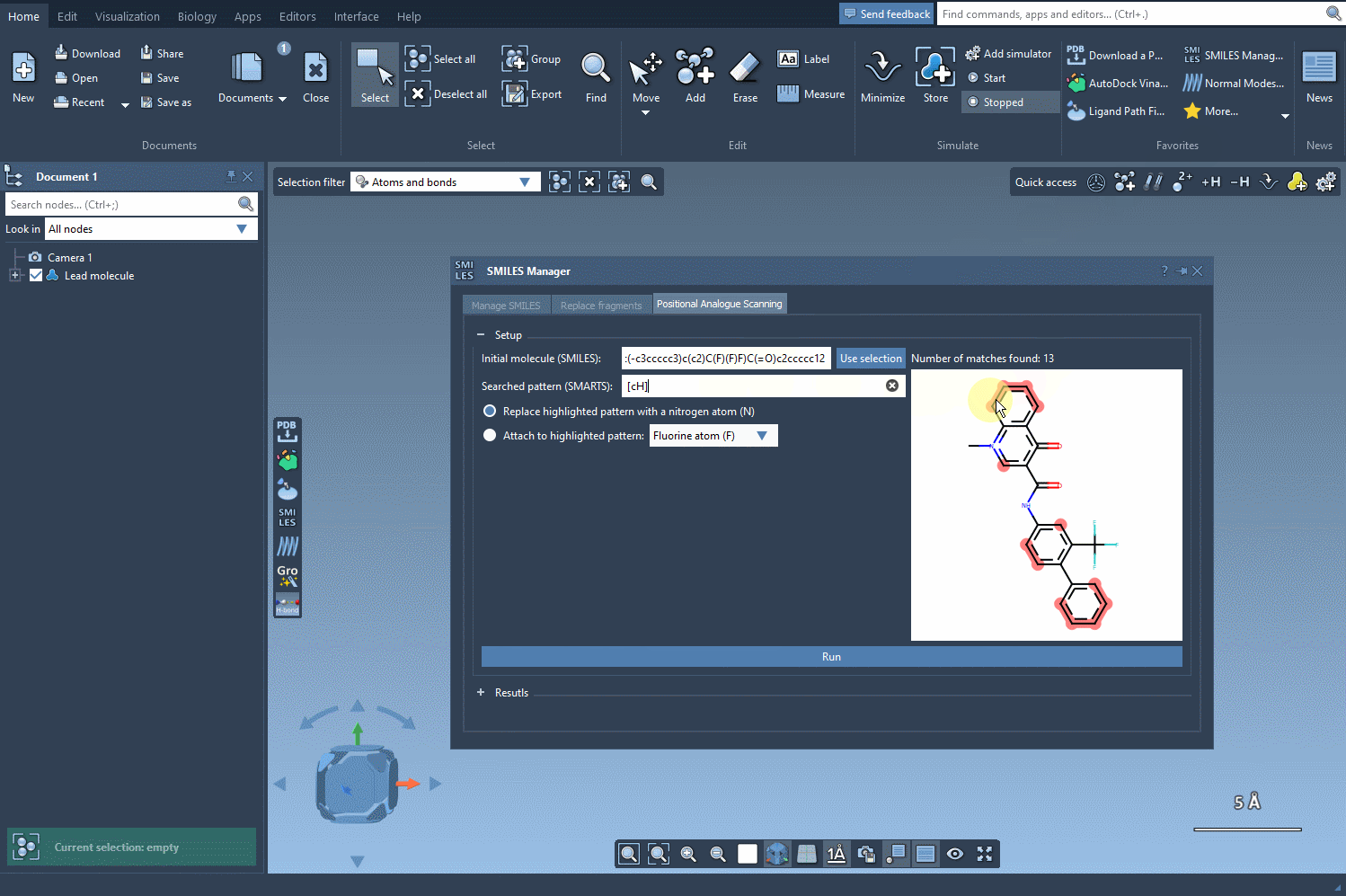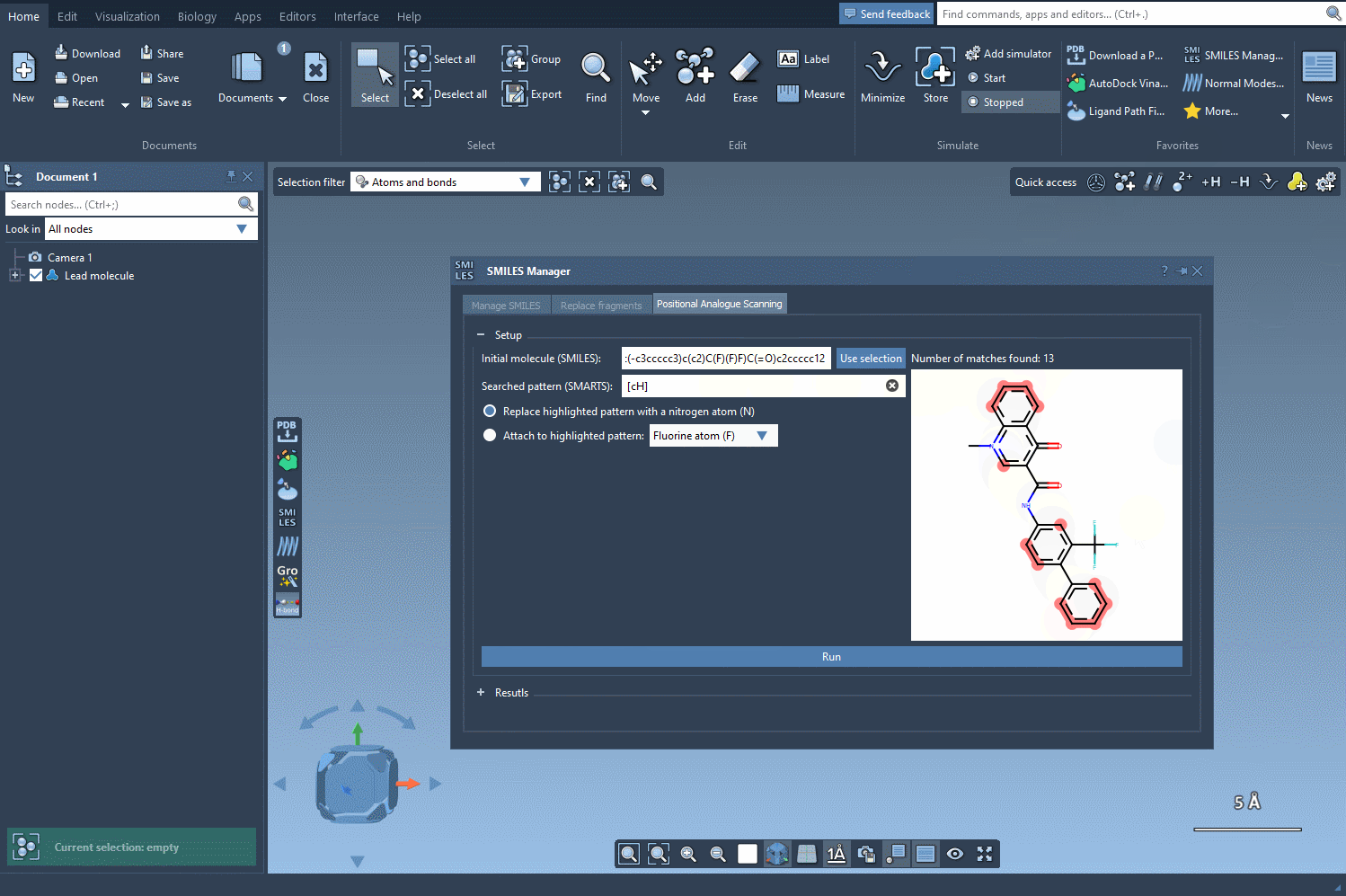
Start by selecting your molecule of interest. If you have the SMILES code, you can enter it directly into the interface. Alternatively, select the molecule in your SAMSON document and click Use selection. It’s that easy to define your starting point.
Target a chemical pattern
Now, define the structural motif where modifications will be introduced. This is done using SMARTS patterns. For instance, you can target aromatic hydrogens using [cH]. When the pattern is detected, it’s highlighted in the molecular structure for visual verification. This makes it easier to see exactly where changes will occur.
Replace or attach groups easily
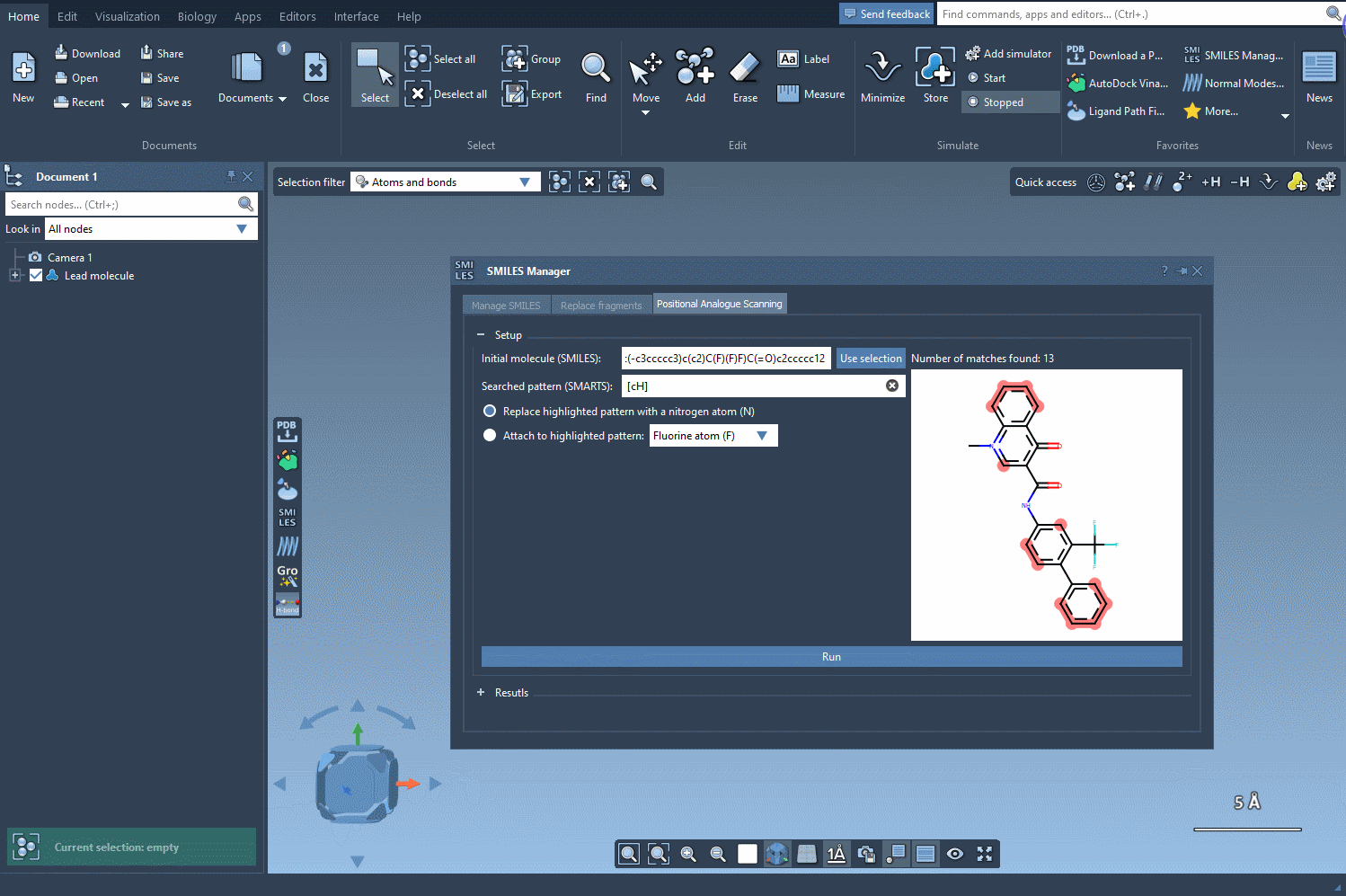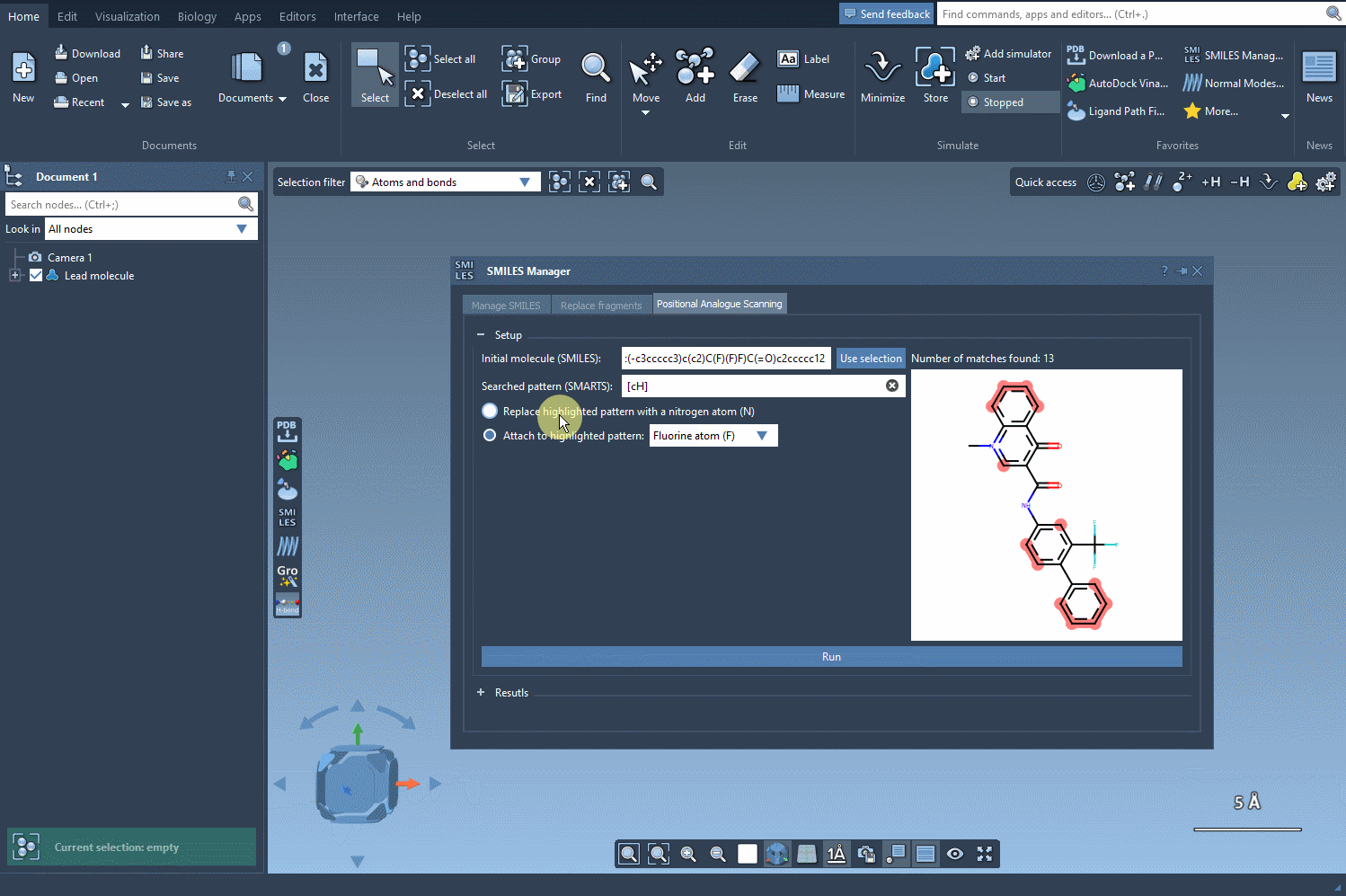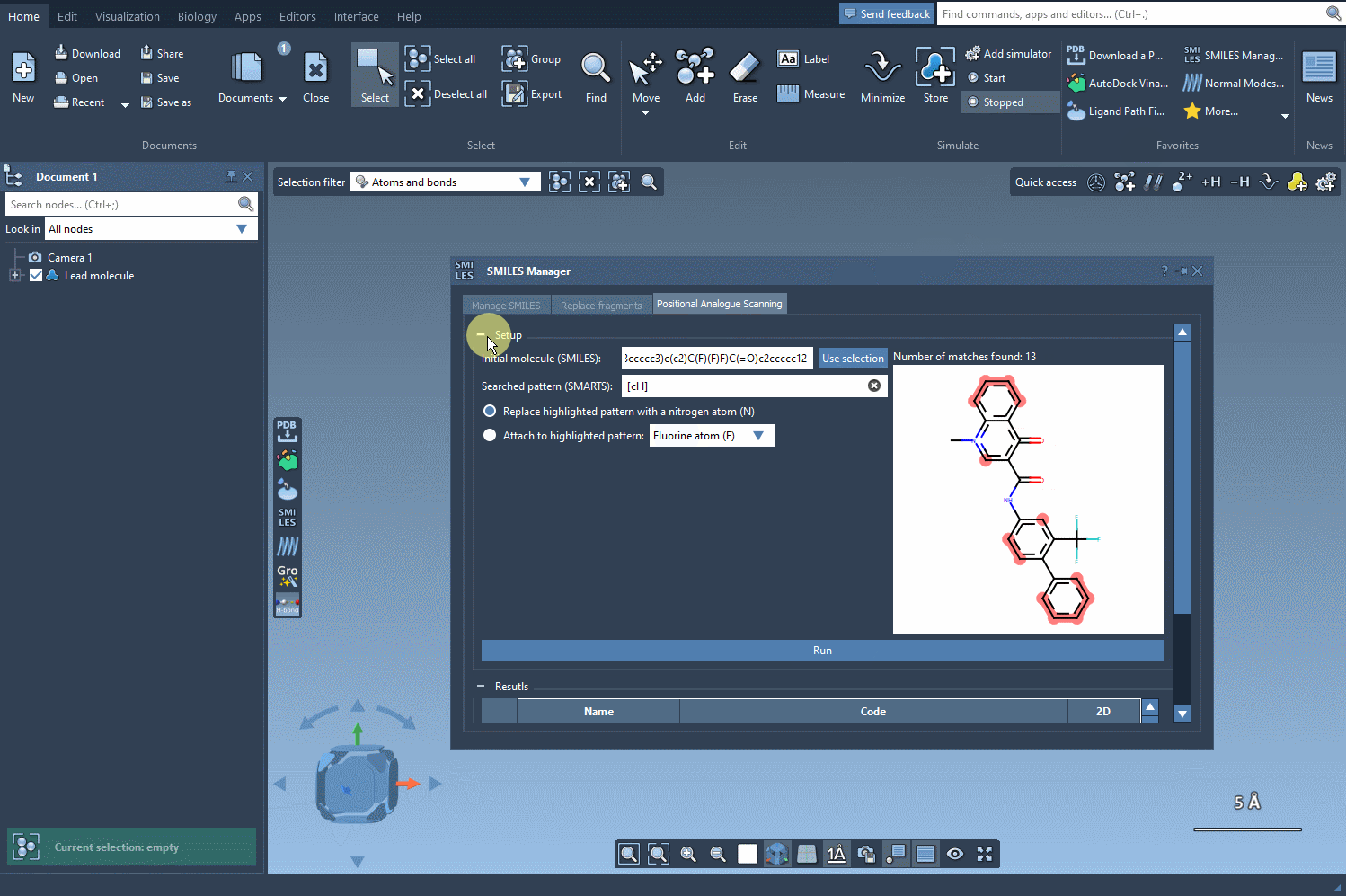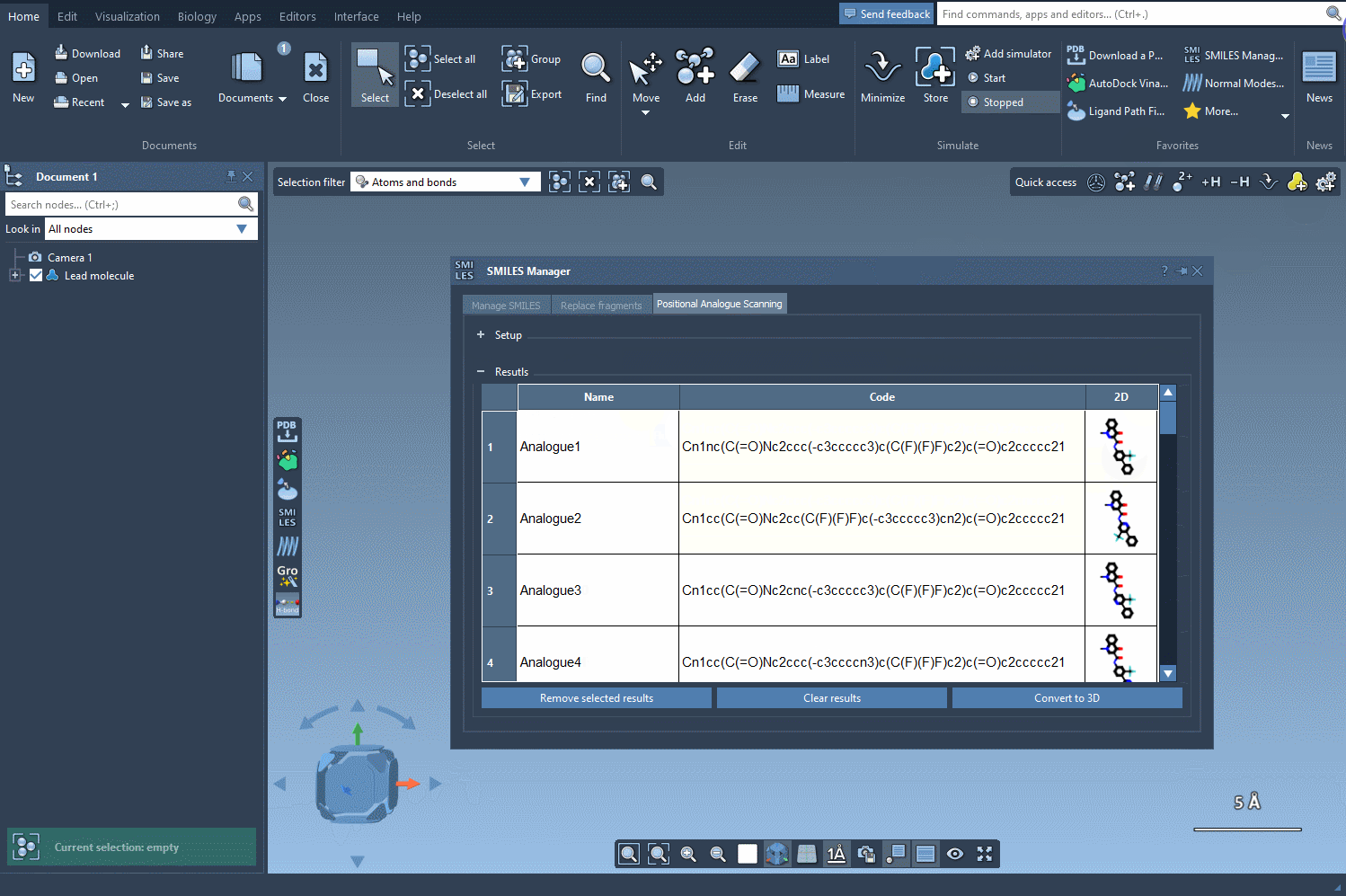
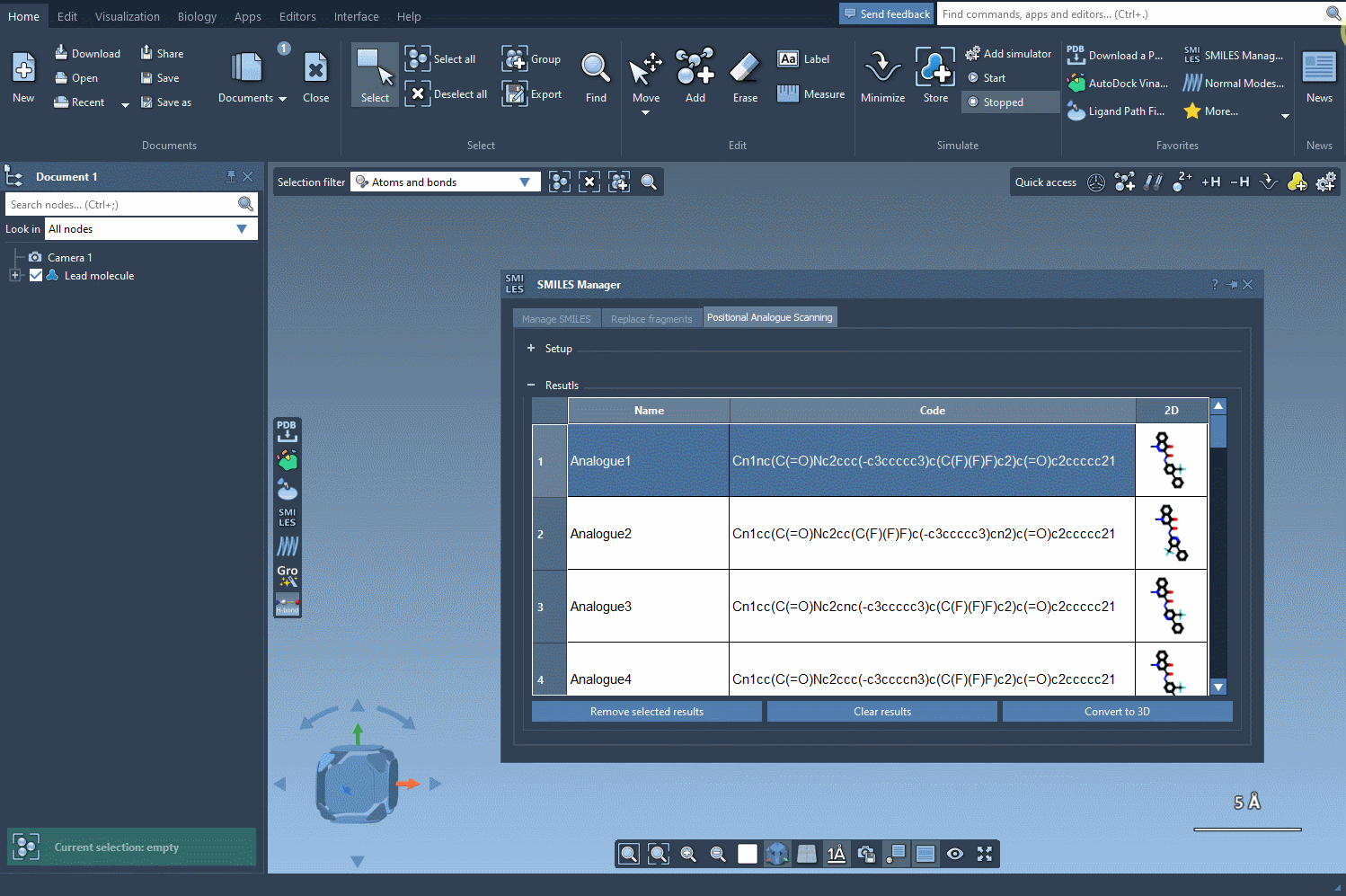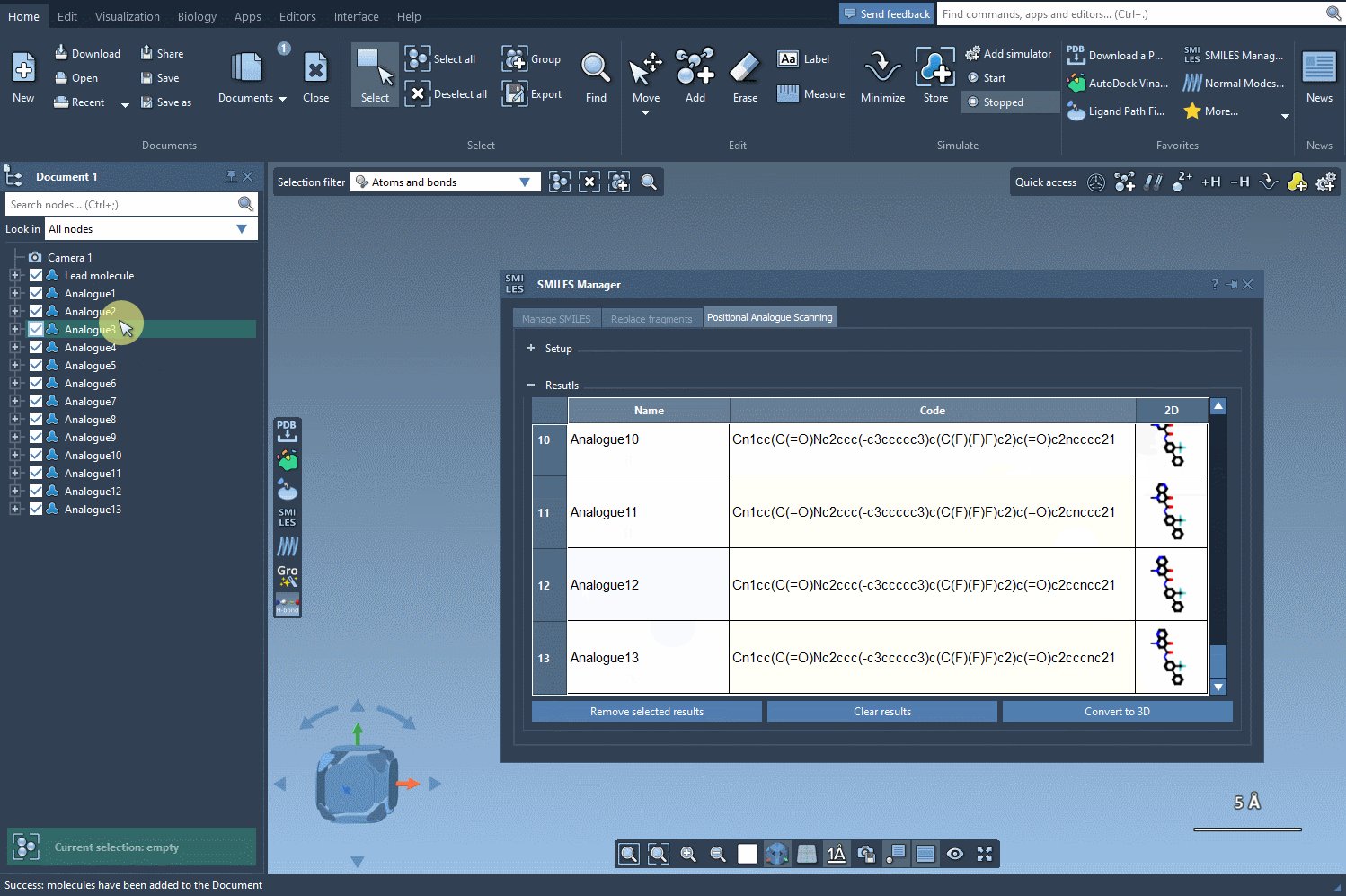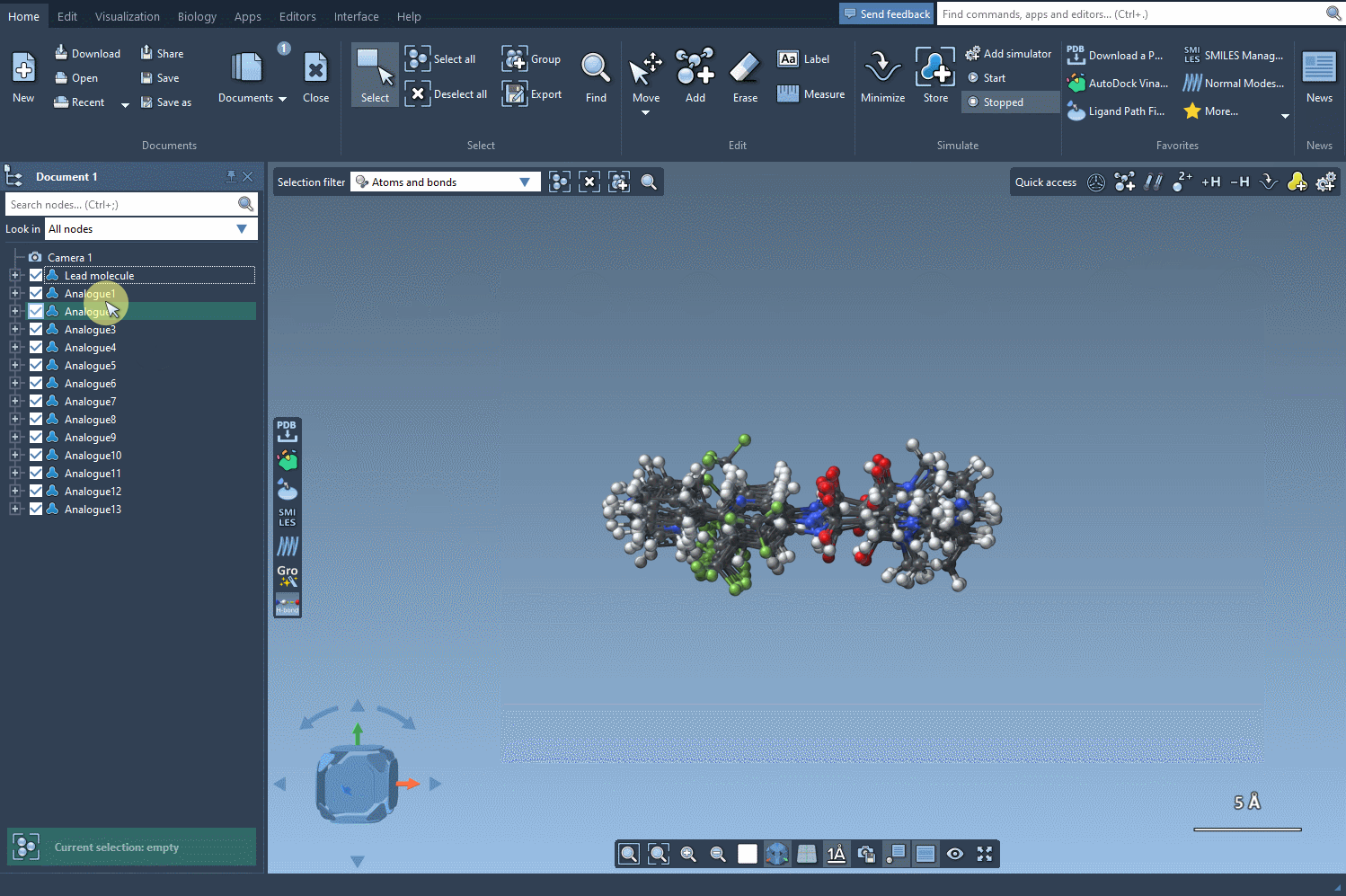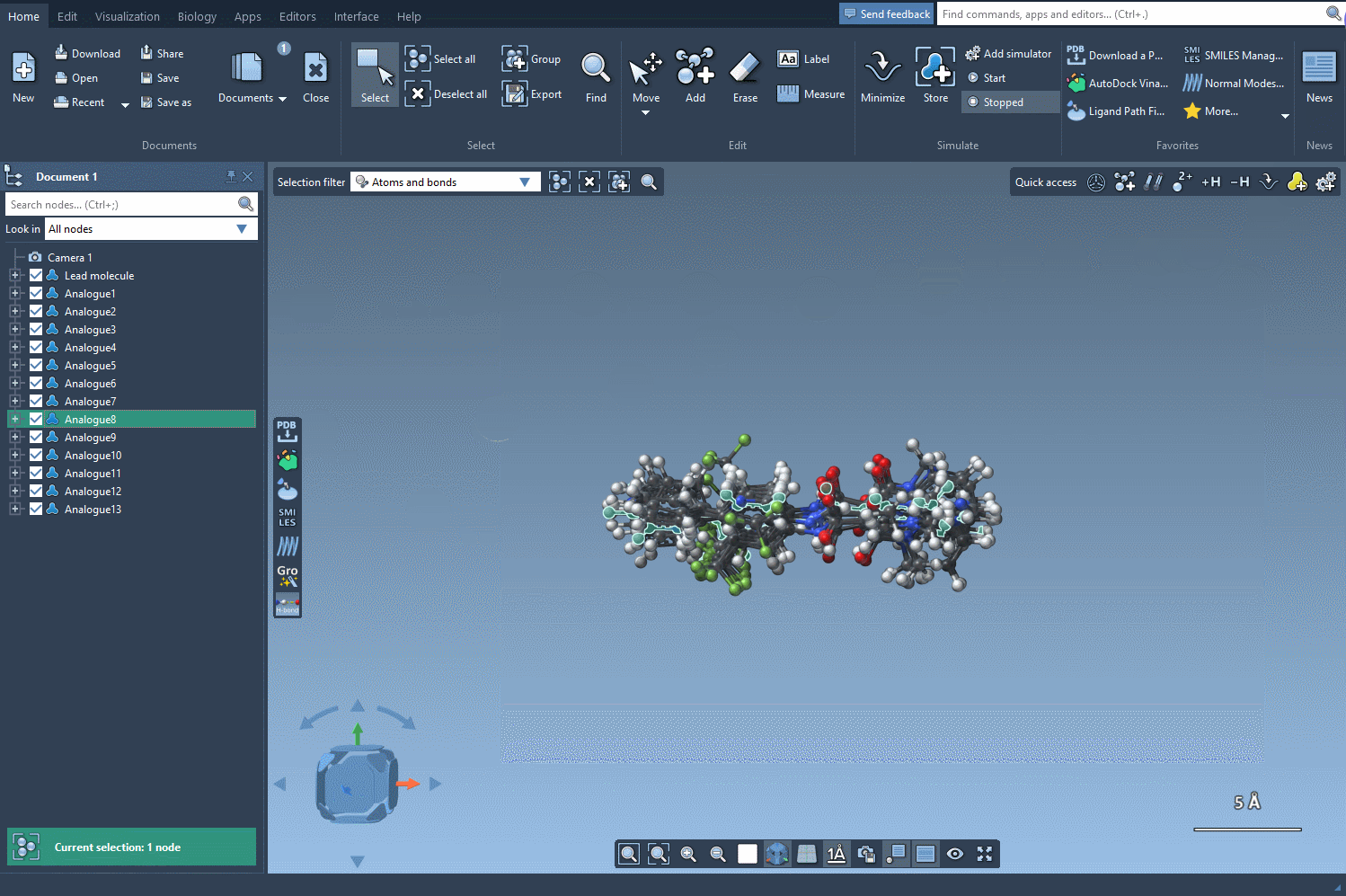
Once you’ve selected the pattern, choose whether to replace it completely (e.g., with a nitrogen atom) or attach a new group (like -F or -CH3). With a single click on the Run button, SMILES Manager generates analogs based on your choices and displays them in the results panel with 2D depictions.
Visualize, analyze, and refine your analogs
Your analogs appear in a table where you can:
- View or hide 2D representations
- Rename or edit the SMILES code directly
- Generate 3D structures with a right-click
- Filter, delete, or refine your working set before proceeding
What’s next?
Once your analogs are ready, you can convert them to 3D and dock them into a target protein using the Autodock Vina Extended extension. This allows you to directly inspect how small changes impact key interactions inside the binding pocket.
Is this for you?
Whether you’re running SAR studies, probing structure-activity relationships, or just exploring ways to improve potency, this SAMSON workflow can eliminate repetitive tasks and help you test ideas faster—visually and interactively.
If you’re interested in replicating this workflow or trying it with your own compounds, check out the full documentation at this page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON here.