





Molecular modeling often involves studying complex energy landscapes to understand transition pathways between states. Whether you are investigating ligand unbinding, protein conformational changes, or reaction mechanisms, obtaining accurate and physically meaningful transition paths can be challenging, especially if your initial path is too rough or lacks precision.
The P-NEB (Parallel Nudged Elastic Band) app in SAMSON is a powerful solution for refining rough paths or sets of conformations into optimized and physically meaningful transitions. Let’s dive into the practical steps of using P-NEB to improve your existing paths and learn how this method can save time while providing precise results.
What is P-NEB?
The Nudged Elastic Band (NEB) method optimizes a series of intermediate structures, or “images,” along a transition pathway. At the same time, it uses spring forces to ensure the states remain evenly distributed, preventing clustering.
The P-NEB app in SAMSON enhances this process by supporting parallel execution, enabling faster performance, particularly when optimizing extensive systems.
When Should You Use P-NEB?
P-NEB is best applied when you already have a rough path or a set of conformations and want to refine them into a more realistic representation of the system. Below are some scenarios where P-NEB becomes indispensable:
- You have multiple structures corresponding to energy minima and need to identify the transition path between them.
- A rough initial trajectory already exists, and you need to refine it for greater accuracy.
- You are investigating ligand unbinding, binding, or other complex molecular interactions requiring optimized energy pathways.
Preparing Your Workspace
To get started with P-NEB, ensure you have:
- The P-NEB Extension installed in SAMSON.
- An initial pathway, such as a rough conformation series or a path node in the Document view.
Additionally, if your goal is to find paths between two relaxed states, you can generate interpolated conformations using SAMSON Extensions such as the Ligand Path Finder.
Applying P-NEB to a Path
Once your initial path is loaded in SAMSON:
- Navigate to Home > Apps > All > P-NEB to open the P-NEB interface.
- Adjust the app settings to tailor the optimization process. For example:
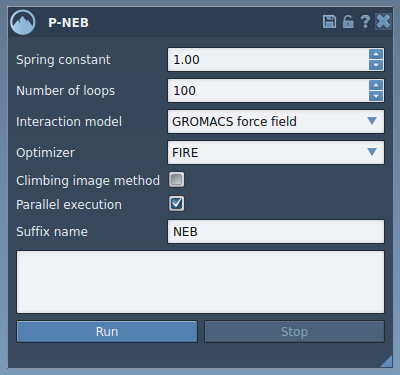
- Set Spring constant to maintain sufficient spacing between path nodes (e.g., 1.00).
- Choose an optimizer like FIRE for efficient energy optimization.
- Select the desired Interaction model, such as “Universal Force Field.”
- Enable Parallel execution for faster computations on multi-core systems.
- Click Run, and confirm the use of existing bonds in the resulting dialog.
- Monitor the optimization progress via the status bar, and review the output path in the Document view upon completion.
Below is a screenshot of the P-NEB interface to guide you as you work:
Why Use P-NEB for Refining Conformations?
You can also use P-NEB directly on a set of conformations, grouping them into a coherent path. However, this approach can take longer compared to refining an existing path. To streamline your workflow, consider combining conformations into a single path first (via Conformation > Create path from conformations in the context menu).
Visualizing the Results
Once the optimization completes, a refined path or set of conformations will appear in the Document view. From here, you can:
- Inspect the energy distribution using the Inspector.
- Animate the path by double-clicking it.
- Export the optimized trajectory for further analysis or simulation workflows.
Here’s an example of the resulting path in the Document view:
Conclusion
Using the P-NEB app in SAMSON helps molecular modelers obtain accurate and physically realistic transition paths in a systematic and efficient way. Whether you’re refining ligand dynamics or protein conformational changes, P-NEB ensures your pathways are refined with precision.
To explore additional optimization techniques or related tools, visit the full documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. Start your journey in molecular design by downloading SAMSON at https://www.samson-connect.net.