Unraveling the Power of Formal Charge Attribute in Structural Models
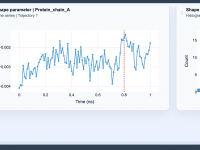
For molecular modelers, one of the most common challenges is finding specific structural models that meet particular criteria—whether it’s about physical properties, chemical behavior, or molecular configurations. For scientists working with charged molecules or ionic interactions, identifying structures with specific…