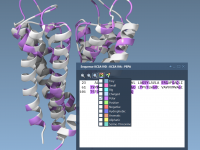
For molecular modelers, one size rarely fits all. Whether it’s visualizing intricate molecular structures, running customized simulations, or crafting specialized computational tools, tailoring software to meet unique requirements can be challenging. This is where SAMSON comes into play with its…