





Author: OneAngstrom
Preserving Your Ideal View with the ‘Hold Camera’ Animation in SAMSON
Streamlining Ligand Preparation with AutoDock Vina Extended in SAMSON
A Closer Look at Predicting Biomolecular Structures with AlphaFold-2 in SAMSON.
Understanding the Opening Motion of the SARS-CoV-2 Spike Protein.

Mastering Visual Presets in Molecular Modeling: A Hands-On Guide
Streamlining Batch Computations in Molecular Dynamics Simulations
Running complex molecular dynamics simulations often involves preparing and computing across multiple molecular systems or conformations. When single simulations are manageable, large-scale batch computations can become time-consuming and prone to errors. If you find yourself facing this challenge, the GROMACS…
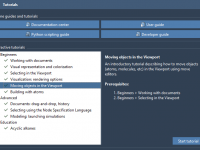
Master Molecular Workflows with SAMSON Interactive Tutorials
For molecular modelers diving into the complex world of computational design, one common challenge is translating theoretical workflows into practical application. This is where SAMSON Interactive Tutorials come into play. These tutorials provide hands-on guidance directly inside the software, making…
Streamline Molecular Modeling: Efficiently Loading Files in SAMSON

Refining Your Molecular System: How to Handle Crystal Waters Outside the Active Site
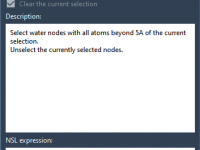
Molecular modelers often face challenges when preparing protein models for simulations, particularly when dealing with crystal waters outside the active site. These water molecules, if left unchecked, can unnecessarily complicate simulations and lead to inefficiencies. However, carefully preserving or removing…