





Author: OneAngstrom
Filter and Explore Your Molecular Visual Models Effectively
Simplify Your Molecular Modeling with SAMSON Extensions.

Streamline Your Molecular Modeling with SAMSON Apps: A Quick Guide
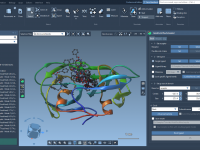
Effortlessly Align Protein Structures and Sequences in SAMSON
For molecular modelers, one common challenge is comparing protein structures and sequences to understand similarities, variations, or functional insights. This process can often feel complex, especially when working with large datasets or intricate structures. SAMSON’s Protein Aligner could dramatically simplify…
Simplify Molecular Modeling with Dark Mode in SAMSON
Effortlessly Convert SMILES Strings into 3D Molecular Structures
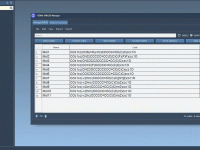
Molecular modelers often encounter the challenge of converting textual molecular representations (SMILES strings) into actionable 3D structures for simulation, analysis, or visualization. If you’ve faced this bottleneck, the SMILES Manager in SAMSON offers a straightforward and effective solution. Powered by…
An Accessible Guide to Segment Attributes in SAMSON’s NSL
Streamlining NVT Equilibration with GROMACS Wizard
Exploring Backbone Attributes in Molecular Modeling
For molecular modelers, efficiently specifying and manipulating structural information is crucial. SAMSON’s Node Specification Language (NSL) provides a powerful framework for accurately defining attributes, especially through the backbone attribute space. Understanding and utilizing backbone attributes can save time and empower…