Author: OneAngstrom
Mastering Molecular Visualizations with the Orbit Camera Animation
Enhancing Molecular Models With the Pulse Animation in SAMSON
Refining Transition Paths in Molecular Modeling: An Introduction to P-NEB
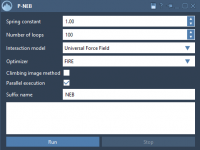
Understanding transitions between molecular states, such as conformational changes or ligand unbinding, is crucial in molecular modeling. However, finding a physically meaningful path between two states can be challenging, especially when dealing with complex energy landscapes. That’s where the Parallel…
Precise Molecular Manipulation with Move Editors in SAMSON
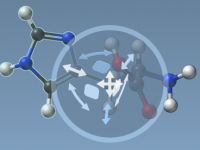
Effortless Bond Creation and Breaking with IM-UFF in SAMSON
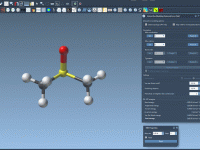
Editing molecular systems interactively can be both fascinating and challenging. Often, modelers need to oversee complex topological changes such as bond creation or breaking, which typically require intricate operations and manual configurations. Enter the Interactive Modeling Universal Force Field (IM-UFF)…
Unlocking the Power of Folder Attributes in SAMSON’s Node Specification Language
Simplify Cloud-Based Molecular Modeling with SAMSON’s Job Manager
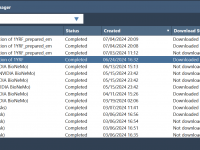
Molecular modeling often involves computationally heavy tasks like protein structure predictions or simulations that require extended computational resources. Managing and organizing these computations efficiently can be challenging. Enter SAMSON’s Job Manager, a tool designed to streamline cloud computations, making them…
Smooth Transitions with the Set Background Animation in SAMSON
For molecular modelers creating presentations or visualizing dynamic processes, ensuring smooth transitions between frames is crucial to maintaining clarity. One potentially frustrating challenge can be presenting complex data with visual consistency, especially when background changes are required. In SAMSON, the…