Author: OneAngstrom
Streamlining Molecular Modeling with AlphaFold-2 in SAMSON
Molecular modeling often hinges on accurate structure predictions to enable simulations, docking studies, and drug discovery exploration. Yet obtaining reliable biomolecular models can feel daunting, especially when juggling different tools for prediction. Here’s how the AlphaFold-2 integration in SAMSON can…
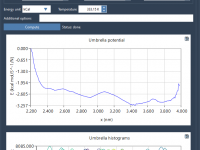
Transform Your Simulation Data into PMF Insights with GROMACS Wizard
Mastering Molecule Attributes in SAMSON’s NSL
Molecular modeling is an essential part of scientific research and design, yet navigating complex molecular datasets can sometimes feel overwhelming. One key challenge is efficiently querying molecular properties to extract meaningful insights. Thankfully, SAMSON’s Node Specification Language (NSL) offers a…
Master Folder-Level Insights with SAMSON’s NSL Attributes
Molecular modelers often find themselves managing complex molecular datasets, trying to streamline workflows and extract specific information efficiently. SAMSON’s Node Specification Language (NSL) offers an invaluable toolset to help modelers refine their queries and navigate data structures efficiently. One particularly…
Understanding Path Attributes in SAMSON’s NSL System
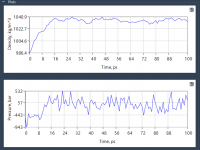
Mastering NPT Equilibration in Molecular Modeling
Master the Preparation of Coarse-Grained Systems in GROMACS Wizard
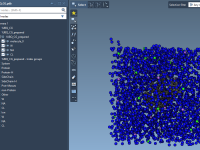
Preparing coarse-grained (CG) systems for molecular dynamics simulations can sometimes be complex and prone to errors, especially when ensuring structural accuracy and compatibility with force fields. Fortunately, the GROMACS Wizard in the SAMSON platform simplifies this process significantly. Below is…
Effortless Molecular Topology Editing with IM-UFF: A Guide
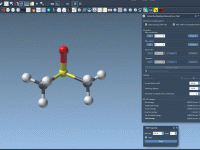
Molecular modelers often face the challenge of smoothly editing molecular topologies while ensuring simulations remain physically meaningful. For instance, breaking or creating bonds, changing bond orders, or adjusting atom typization can cause significant disruptions in a traditional modeling workflow. This…