Mastering Interactive Simulations in SAMSON: A Step-by-Step Guide

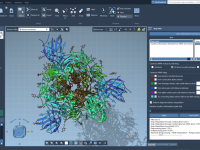
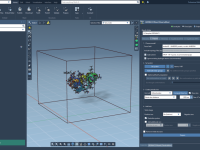
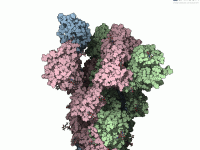
For molecular modelers, one of the most challenging tasks is bridging the gap between theoretical models and dynamic, interactive simulations. Whether you’re optimizing geometries, analyzing systems under different forces, or building molecular pathways, achieving accuracy and interactivity can be daunting.…