Simplifying Molecular Geometry Optimization with the FIRE Minimizer

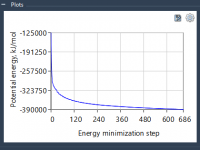
Molecular modelers often face a critical challenge: preparing stable and realistic molecular geometries for simulations or molecular design workflows. Whether you’re cleaning up a structure before running simulations or optimizing geometries during interactive modeling, time and accuracy matter. This is…