





Author: OneAngstrom
Streamline Molecular Visualization with the ‘Shown’ Animation Effect
Streamlining Molecular Models with SAMSON’s Pattern Editors
Creating complex, nanoscale molecular architectures is often a challenge for molecular modelers due to the precision and repetitive tasks involved. Whether you’re designing nanotubes, arranging biomolecular assemblies, or developing material prototypes, SAMSON’s Pattern Editors can transform what often feels like…
Easily Create Molecular Analogues with Positional Analogue Scanning in SAMSON
Ensure Reliable Simulations: A Step-by-Step Guide to Protein Preparation in SAMSON.
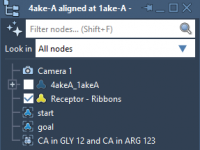
Exploring Chain IDs for Precise Molecular Modeling
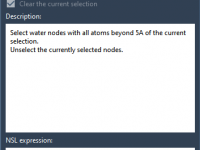
Efficiently Removing Unnecessary Waters in Molecular Simulations
Streamlining Transition Paths with the Parallel Nudged Elastic Band Method
Mastering Folder Attributes in SAMSON for Effective Molecular Modeling
Enhance Transition Pathways with the Nudged Elastic Band Method
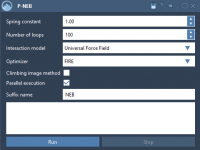
Modeling molecular transitions often involves determining accurate pathways between two states—say, a protein-ligand binding event or a conformational change of a macromolecule. However, the challenge lies in refining these paths to obtain physically meaningful, optimized transitions that can reveal key…