





Author: OneAngstrom
Mastering Conformation Attributes for Better Molecular Modeling.
Effortless Nanotube Modeling with SAMSON’s Nanotube Creator

For molecular modelers and nanoscientists, constructing precise carbon nanotube (CNT) models can be a daunting task. Whether you’re designing CNT-based sensors, exploring electronic properties, or conducting molecular simulations, having the right tools can make all the difference. Enter SAMSON’s Nanotube…
Mastering Light Attributes in Molecular Design with SAMSON
For molecular modelers, controlling visualization and interactions within a complex molecular design environment can significantly streamline their workflow. SAMSON’s integrative molecular design platform simplifies this through its light attributes, an essential toolset designed specifically for managing light nodes—elements that represent…
Mastering Visibility in Molecular Models with the ‘Show’ Animation
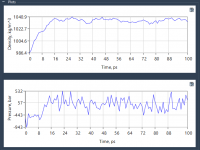
Streamline Your Umbrella Sampling Workflow with GROMACS Wizard.
Why SAMSON Doesn’t Require Admin Rights—and Why That Matters
When it comes to molecular modeling tools, many professionals and students face a significant hurdle: installation challenges on restricted systems. Laboratories, universities, and companies often impose strict administrative controls, making it frustrating to install software that requires admin privileges. SAMSON,…
Streamlining Molecular Visualization with SAMSON’s Visual Presets
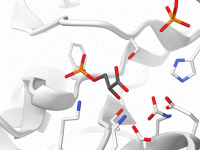
One of the significant challenges molecular modelers face is the efficient visualization of complex molecular systems. Whether analyzing protein-ligand interactions or highlighting specific structural elements, having a streamlined, intuitive way to create meaningful visualizations is essential. This is where SAMSON’s…
Enhance Your Molecular Simulations with Custom Index Groups in GROMACS Wizard
Simplifying Molecular Animations: A Guide to Animation Attributes in SAMSON
For molecular modelers, efficiently annotating and managing animations can significantly enhance workflows, especially when dealing with complex systems. The Animation Attributes in SAMSON’s Node Specification Language (NSL) provide tools to optimize how molecular animations are defined, interacted with, and visualized.…