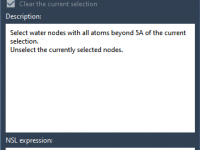
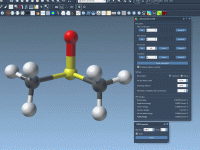
Making the Most of Property Model Attributes in Molecular Design
When modeling molecular systems, it’s crucial to keep track of node attributes for better control, organization, and customization. This is particularly relevant in SAMSON, the powerful integrative molecular design platform, where Property Model Attributes can significantly enhance your workflow by…