Author: OneAngstrom
What If You Could Just Talk to Your Molecular Modeling Platform?
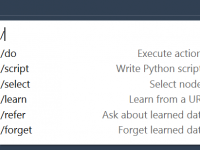
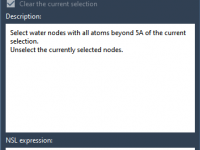
Many molecular modelers spend significant time navigating menus, remembering shortcuts, and writing scripts to perform everyday tasks: visualizing surfaces, modifying representations, or selecting atom groups. While flexibility is appreciated, the friction can add up—especially when switching between modeling tools and…
When It Matters: How to Use Your Own Version of GROMACS in SAMSON
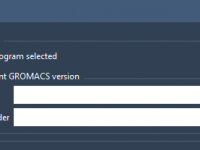
Switching between different versions of GROMACS isn’t just a technical preference—it’s often a necessity for molecular modelers. Whether for ensuring reproducibility, taking advantage of specific performance improvements, or simply using a cluster’s precompiled GROMACS, being able to control the version…