Customizing Bond Graphs in Your Molecular Simulations
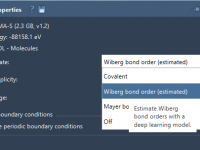
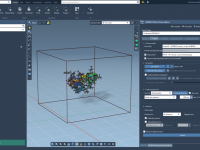
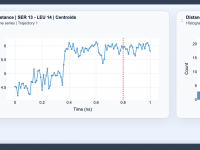
For molecular modelers, refining and understanding bond structures during simulations can be a challenging task. Whether you’re optimizing molecular geometries, exploring chemical reactivity, or examining material properties, the way bonds are updated can significantly affect your insights. This blog post…