Author: OneAngstrom
Mastering Protein Path Finder: Essential Setup for Smooth Pathway Analysis
Protein conformational transitions are at the heart of many biological processes, but modeling these transitions can often feel like juggling multiple complexities. One common challenge molecular modelers face is ensuring their molecular systems are adequately prepared before running computational pathways—where…
Mastering Molecular Modeling with SAMSON’s Document View
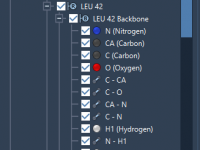
Mastering File Import in SAMSON: A Guide for Molecular Modelers
Mastering the Setup of UMA Force Field in SAMSON
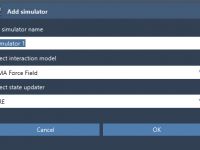
Setting up an effective molecular simulation can feel overwhelming, particularly if you’re working with advanced machine-learning tools. Many molecular modelers struggle with efficiently setting up workflows that combine precision and speed. One highly effective tool for energy and force evaluation…