When crafting molecular animations, researchers often want to highlight movement in one part of a system while keeping another part still for clarity or comparison. For example, during a protein-ligand docking animation, you might want the protein to remain static…
For many molecular modelers, one recurring challenge is the lack of flexible scripting capabilities in their software tools. Whether the goal is to extract custom properties, automate repetitive tasks, or prototype an analysis workflow, scripting support can often make the…
Understanding how a ligand dissociates from a protein binding pocket is a key question in many areas of molecular modeling and drug design. Once you’ve found potential unbinding paths, the next practical challenge is making sense of them—where does the…
For structural biologists and molecular modelers, the frustration of navigating complex macromolecular systems is all too familiar. You often know the residue or chain you need to focus on—but shifting through a crowded 3D visualization can be tedious and inefficient.…
When working on complex molecular modeling tasks, Python scripting can save hours of repetitive work. However, switching between external editors, terminals, and visualization platforms often breaks your workflow and makes it harder to test and debug scripts on the fly.…
One of the common challenges molecular modelers face is managing complex scenes filled with numerous nodes — atoms, molecules, property models, and materials. A cluttered viewport can slow down analysis, impact rendering performance, and simply make it harder to focus.…
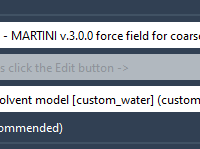
When preparing coarse-grained (CG) molecular dynamics simulations with the MARTINI force field, many modelers spend time double-checking whether they’ve configured the correct parameters and files. One small but frequent pitfall is using a CG topology—often from Martinize2—but forgetting to assign…
When presenting a molecular simulation or model to an audience, timing is everything. You may want to highlight a key step in a mechanism, pause for discussion, or transition smoothly between ‘slides’ of your demonstration. But how do you break…
One of the most common frustrations molecular modelers face is being unable to save their work in the right format. Whether you’re preparing to submit a structure to a database, running simulations in an external program, or simply sharing an…
Exploring how small changes in your molecules impact their behavior is a key activity in molecular modeling, especially during hit expansion or lead optimization. But trying out dozens of variations by hand? That gets tedious quickly. This is where the…