Category: Uncategorized
No Admin Rights? No Problem: Installing SAMSON Without Elevated Privileges
How to Quickly Find Molecular Paths by Number of Atoms in SAMSON
Precise Atom Selection Without Coding
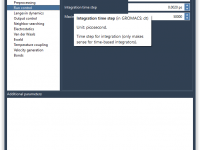
Customizing Your MDP Files in SAMSON’s GROMACS Wizard
Export Trajectories of Selected Atoms Along Ligand Unbinding Paths
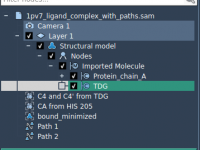
Understanding molecular interactions often requires not just static snapshots of a system, but watching how molecules move—especially when investigating mechanisms like ligand unbinding or conformational transitions. One common challenge for computational chemists and molecular modelers is the need to export…
A Precise Way to Rotate Molecules in SAMSON: Rotation Snapping Made Easy
Label Management in Molecular Models: Make Your Viewport Cleaner and Smarter
Metallic or Glassy? How to Control Molecular Materials in SAMSON with Cycles
Make Molecules Appear When You Need Them: Mastering the Show Animation in SAMSON
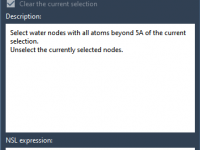
No Admin Rights? No Problem: Installing SAMSON Without Elevated Privileges
How to Quickly Find Molecular Paths by Number of Atoms in SAMSON
Precise Atom Selection Without Coding

Customizing Your MDP Files in SAMSON’s GROMACS Wizard
Export Trajectories of Selected Atoms Along Ligand Unbinding Paths
Understanding molecular interactions often requires not just static snapshots of a system, but watching how molecules move—especially when investigating mechanisms like ligand unbinding or conformational transitions. One common challenge for computational chemists and molecular modelers is the need to export…