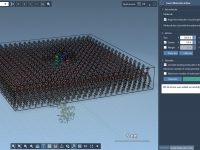
Building realistic lipid environments around membrane proteins can be challenging and time-consuming. Ensuring proper orientation of the molecule, avoiding atom overlaps, adding buffer space… these steps often involve a patchwork of tools and manual adjustments that take hours—even before you…
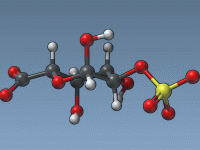
When working with complex molecular systems, it is often necessary to repeatedly check specific distances, angles or torsion angles between atoms. However, a common frustration among molecular modelers is the need to re-measure these values after every view change or…
If you’re a molecular modeler juggling multiple tools, supplementary data, scripts, or even presentation figures, you’ve likely experienced the frustration of losing track of crucial files. Whether you’re preparing a publication, sharing a molecular model with collaborators, or switching between…
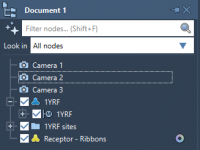
A common pain point for molecular modelers is the need to quickly select and manipulate secondary structure elements—like alpha helices, beta sheets, and loops—in large biomolecular systems. Whether you’re preparing a visualization, performing a simulation setup, or designing mutations, manually…
For molecular modelers working with complex systems, keeping track of annotations within a project can be just as crucial as modeling structures and reactions. Whether you’re leaving comments for collaborators or marking specific components for further analysis, notes in SAMSON…
When presenting molecular designs or creating educational content, timing and visibility can make a big difference. Instead of overwhelming viewers with full molecular structures displayed at once, imagine revealing atoms and bonds gradually—highlighting the assembly or focus of a system…
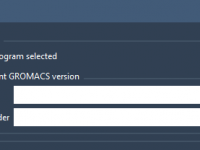
Molecular modelers working with GROMACS often rely on specific versions of the simulation software, either for reproducibility or to take advantage of local custom builds. However, many tools that simplify the setup of molecular dynamics simulations don’t make it easy…
When working on complex molecular systems, visual clarity is vital. Whether you’re zooming in on a protein-ligand interaction or looking at the overall structure of a macromolecular complex, being able to shift perspectives quickly makes a difference. Fortunately, SAMSON offers…
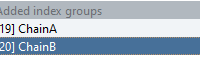
Molecular modelers often face a common hurdle when setting up pulling simulations using GROMACS: how to define custom index groups that don’t appear in the default list. Whether it’s pulling a specific protein chain or restraining a certain region of…
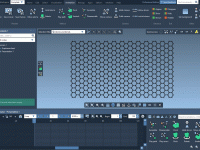
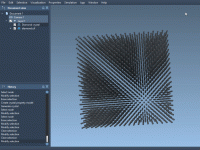
One of the challenges in simulating real materials is going beyond the ideal crystal structure. In physical reality, materials often contain defects, which can completely alter their properties — from electronic behavior to mechanical resistance. Fortunately, the Crystal Creator extension…