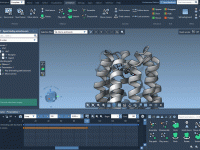
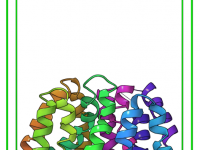
If you’ve ever imported a crystal structure and wondered why it doesn’t reveal the atomic layout you expected, you’re not alone. Many molecular modelers load a crystal, spin it around a few times, and still struggle to see the packing…
One of the most common questions from molecular modelers preparing visual presentations or simulations is: How do I keep track of where atoms move during an animation? Whether you’re demonstrating a molecular docking sequence, a structural assembly, or the results…
If you’ve ever tried to manually click your way through a complex molecular model just to select a few specific atoms, chains, or residues, you know how tedious and error-prone it can be. The process becomes even harder when dealing…
Preparing molecular animations is a frequent task in structural biology, medicinal chemistry, and computational modeling. One challenge that often arises is how to visually present molecules as they dock into a receptor, making this interaction clear, educational, and even aesthetically…
When building or analyzing molecular models, selecting relevant structural components efficiently can be a tedious process. For example, you may want to isolate all molecular paths that contain a certain number of atoms—say, to study backbone flexibility in oligomers, filter…
Filtering molecular structures by size can speed up your workflow significantly when working with large and complex models. If you’ve ever struggled to isolate paths with a specific number of atoms in SAMSON, the Node Specification Language (NSL) offers a…
If you’re a molecular modeler, you’re probably all too familiar with repetitive tasks: loading structures, setting up visual styles, preparing systems, analyzing trajectories, and more. These operations can quickly become tedious, especially when they need to be performed multiple times…
When working with complex molecular trajectories, a common challenge for researchers and molecular modelers is maintaining visual focus on a specific region of the system over time. Imagine you’re simulating a protein-ligand binding event or monitoring the conformational dynamics of…
Finding ligand unbinding pathways through protein tunnels is a common need in molecular modeling. But did you know that the way you define the sampling region can significantly influence the results? In this blog post, we focus on a critical…
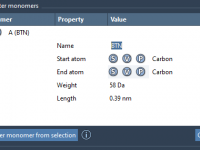
If you’re working with custom polymers, one of the most repetitive (and error-prone) steps in your workflow might be building complex chains from fragments you designed. Whether you’re researching synthetic polymers or developing molecular scaffolds for prediction models, you likely…