Faster Molecular Illustrations with Visual Presets in SAMSON
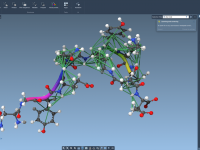
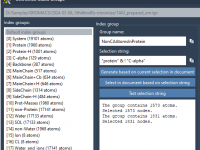
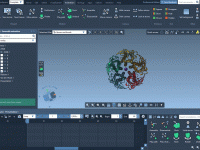
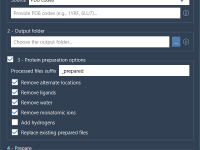
For molecular modelers juggling complex structures, creating beautiful and informative molecular visualizations can feel like a time-consuming process. Manually setting colors, choosing the right visual models, and fine-tuning rendering effects is often repetitive and error-prone, especially under tight publication or…