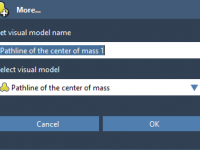
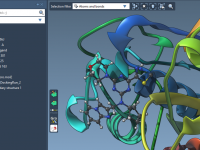
Molecular modelers often need to understand how small molecules (ligands) move in and out of binding pockets in proteins. Whether you’re studying unbinding mechanisms for drug design or analyzing ligand diffusion paths through protein tunnels, visualizing these motions helps make…
For molecular modelers, one of the earliest — and often most frustrating — obstacles in a project is simply getting the data in. Files from different sources can be in various formats, and not all software handles them equally. If…
Molecular dynamics simulations often take time, especially when running production-scale simulations with thousands of atoms. One common frustration among molecular modelers is that running such simulations can lock up their software or prevent them from continuing to work, forcing them…
Covalent docking is an essential technique for drug discovery projects targeting enzymes and other proteins with reactive residues. However, setting up a covalent docking experiment can be time-consuming and error-prone, especially if you need to adjust bond orders, assign atom…
If you’ve ever tried to edit a whole group of atoms in your molecular model—maybe to reposition a residue without disturbing the relative geometry of its atoms—you might have found it tricky to avoid collapsing everything into a single point.…
Sharing molecular modeling workflows with colleagues or students often involves zipping together input files, scripts, images, and documentation — and hoping they preserve the folder structure when unpacking. But what if you could combine everything — models, scripts, data, and…
One of the common frustrations in protein-protein docking is the large number of false positives that arise from unconstrained searches. Molecular modelers often spend excessive time manually filtering docking results, only to realize that many of the proposed orientations are…
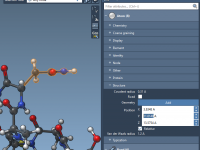
When working on complex molecular systems, one common problem modelers face is the need to edit properties of multiple entities—such as atoms—simultaneously. Whether you’re adjusting atomic positions, changing atom types, or reviewing structural attributes, doing it one atom at a…
When working with complex molecular systems in SAMSON, things can get quite crowded. You may have multiple property models attached to atoms, molecules, or higher-level structures, and you just want to operate on a specific subset—like only those that are…
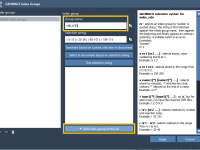
When working with molecular systems in GROMACS, detailed analysis and advanced simulation setups such as umbrella sampling or pulling simulations often require specific selections of atoms or residues. These selections are managed using index groups. GROMACS automatically generates standard index…