If you’ve ever tried to move a group of atoms in your molecular model, only to realize that they’re no longer in the same geometric arrangement, you’re not alone. Many molecular modelers face this issue when managing atomic coordinates for…
Anyone who has worked with complex molecular systems knows that managing atomic structures, ligands, simulations, and annotations can quickly become overwhelming. As molecular modelers, we often lose precious time searching for that one piece of structural data buried deep in…
One of the most common sources of errors during molecular dynamics simulations happens right at the start: providing the wrong input file. Whether due to typos, incorrect paths, or confusion between file types, this early mistake can become a frustrating…
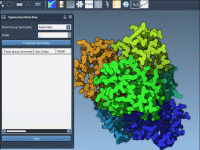
Automatic symmetry detection in molecular structures can be a huge time-saver. But if you’re working with large biological assemblies, such as viral capsids or protein complexes with ambiguous layouts, automated tools might suggest several plausible symmetry groups. For structural biologists…
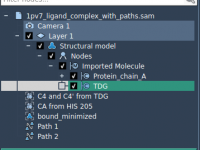
As your molecular models grow bigger and more complex, staying organized becomes essential. Whether you’re aligning protein structures, preparing ligands, or scripting simulations, you’re likely juggling multiple structures, tools, and settings at once. This is especially true when working on…
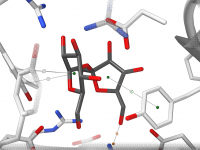
Have you ever wanted to extract ligand motion data for free energy profiling, visualize exit pathways, or selectively track atoms through molecular pathways? Exporting atom trajectories along paths is a frequent but often tedious task for molecular modelers—especially when customizing…
When preparing molecular presentations or animations in SAMSON, a common challenge is transitioning smoothly from invisible to visible structural elements. Rather than having parts of your molecular model snap instantly into view, a progressive appearance helps direct attention and improves…
Exploring large molecular systems, such as proteins or complexes, can often feel like peering into a dense forest—too much to see at once and no clear view of what’s inside. Whether you’re investigating binding sites or preparing images for presentations,…
If you’ve ever tried to animate a molecular simulation and watched your region of interest drift out of frame, you’re not alone. This is a common challenge in molecular modeling when dealing with complex systems—especially when atoms of interest move…
When dealing with complex molecular systems in SAMSON, it can be challenging to quickly isolate groups of nodes—such as atoms, residues, or molecules—based on specific properties. If you’re spending too much time visually scanning your models or writing repetitive selection…