





Category: Uncategorized
Avoid GRO File Confusion: Smarter Input Management with GROMACS Wizard
Quickly Filter Molecular Animation Nodes by Visibility in SAMSON
Tired of Static Molecules? How to Bring Your Molecular Systems to Life for Presentations and Publications
Creating compelling molecular animations can be time-consuming — especially when shifting between research tools and video editing software. Molecular modelers often face the challenge of clearly explaining dynamic phenomena such as docking, conformational changes, or assembly mechanisms. Static images often…
Choosing the Right Discrete Color Palette for Molecular Structures
Keeping Molecular Projects Self-Contained with Embedded Files in SAMSON
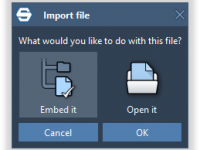
Collaborative molecular modeling projects often involve more than just structural data: Python scripts, research articles, experimental data, machine learning models, and various other resources contribute to replicability and transparency. But organizing and sharing these assets alongside complex molecular systems tends…
When You Know Better: Choosing the Expected Symmetry of Your Molecular Assembly

For many molecular modeling tasks, especially those involving large biological assemblies like viral capsids or protein complexes, understanding molecular symmetry can save time and reduce complexity. But automatic symmetry detection isn’t perfect—sometimes, you already know the answer, and it’s best…
Easily Create Molecular Flybys Without Touching a Timeline
Efficiently Switch Between Molecular Views Using Multiple Cameras in SAMSON

When working on complex molecular structures in SAMSON, navigating efficiently between different viewpoints can significantly improve your productivity and understanding of the system. Whether you’re analyzing binding pockets, exploring lattice arrangements, or reporting structure-function relationships, switching views without hassle is…
Easier Group Selection During MD: Creating Custom GROMACS Index Groups in SAMSON
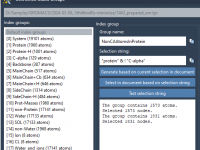
Molecular dynamics (MD) simulations often require targeted control or analysis of specific parts of a structure—for example, calculating forces on side chains, analyzing water clusters, or applying constraints to ligand atoms. GROMACS automatically generates standard atom groups (like Protein, Water,…

Avoid GRO File Confusion: Smarter Input Management with GROMACS Wizard
Quickly Filter Molecular Animation Nodes by Visibility in SAMSON
Tired of Static Molecules? How to Bring Your Molecular Systems to Life for Presentations and Publications
Creating compelling molecular animations can be time-consuming — especially when shifting between research tools and video editing software. Molecular modelers often face the challenge of clearly explaining dynamic phenomena such as docking, conformational changes, or assembly mechanisms. Static images often…
Choosing the Right Discrete Color Palette for Molecular Structures
Keeping Molecular Projects Self-Contained with Embedded Files in SAMSON
Collaborative molecular modeling projects often involve more than just structural data: Python scripts, research articles, experimental data, machine learning models, and various other resources contribute to replicability and transparency. But organizing and sharing these assets alongside complex molecular systems tends…
When You Know Better: Choosing the Expected Symmetry of Your Molecular Assembly
For many molecular modeling tasks, especially those involving large biological assemblies like viral capsids or protein complexes, understanding molecular symmetry can save time and reduce complexity. But automatic symmetry detection isn’t perfect—sometimes, you already know the answer, and it’s best…
Easily Create Molecular Flybys Without Touching a Timeline

Efficiently Switch Between Molecular Views Using Multiple Cameras in SAMSON
When working on complex molecular structures in SAMSON, navigating efficiently between different viewpoints can significantly improve your productivity and understanding of the system. Whether you’re analyzing binding pockets, exploring lattice arrangements, or reporting structure-function relationships, switching views without hassle is…
Easier Group Selection During MD: Creating Custom GROMACS Index Groups in SAMSON
Molecular dynamics (MD) simulations often require targeted control or analysis of specific parts of a structure—for example, calculating forces on side chains, analyzing water clusters, or applying constraints to ligand atoms. GROMACS automatically generates standard atom groups (like Protein, Water,…