Category: Uncategorized
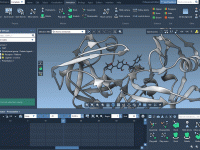
Let Your Molecules Fade Out Gracefully with the Disappear Animation
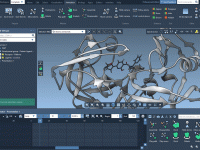
In molecular modeling, clarity is key. Whether you’re presenting a simulation, building an educational video, or simply exploring complex biomolecular structures, drawing attention to specific components while minimizing distractions can greatly improve communication. One common challenge modelers face is how…
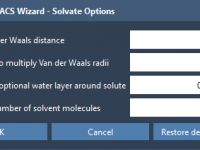
Avoiding Solvent Clashes in Coarse-Grained Simulations with GROMACS Wizard
Quickly Select Aromatic Carbons (and More) with Atom Attributes in NSL
Avoid Repeating Simulations by Recording Atom Paths in SAMSON
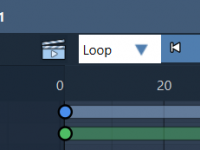
When working on molecular simulations or structural rearrangements, animation workflows can become repetitive and time-consuming. A common challenge for molecular modelers is ensuring that once they achieve the right animation involving atom movements—for example, docking, simulated dynamics, or manual manipulation—they…
Making Molecules Disappear: A Visual Trick for Clearer Presentations
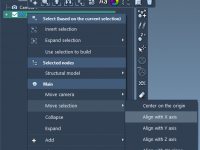
A Practical Guide to Aligning Molecular Structures in SAMSON
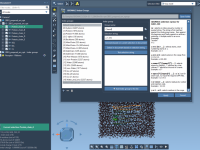
From 2D to 3D: Instantly Visualize Analogs with the SMILES Manager
Creating Custom Index Groups for COM Pulling in GROMACS Wizard
Filtering Molecular Conformations by Atom Count in SAMSON
Let Your Molecules Fade Out Gracefully with the Disappear Animation
In molecular modeling, clarity is key. Whether you’re presenting a simulation, building an educational video, or simply exploring complex biomolecular structures, drawing attention to specific components while minimizing distractions can greatly improve communication. One common challenge modelers face is how…
Avoiding Solvent Clashes in Coarse-Grained Simulations with GROMACS Wizard
Quickly Select Aromatic Carbons (and More) with Atom Attributes in NSL
Avoid Repeating Simulations by Recording Atom Paths in SAMSON
When working on molecular simulations or structural rearrangements, animation workflows can become repetitive and time-consuming. A common challenge for molecular modelers is ensuring that once they achieve the right animation involving atom movements—for example, docking, simulated dynamics, or manual manipulation—they…