Creating molecular animations often comes with a simple but overlooked challenge: making a structure appear clearly and at the right time without visual noise. If you've ever tried to animate molecular models and show a molecule transitioning into the scene,…
When preparing a molecular animation for a presentation, publication, or educational material, a common challenge many researchers face is managing what the viewer sees—and what they don’t. Uncluttered visuals lead to greater clarity, but temporary removal and reappearance of molecular…
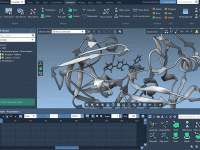
When working on large molecular systems, especially in collaborations or when re-opening older projects, it’s surprisingly common to be puzzled by missing visual elements that should be there. Whether it’s a surface view, a label, or an entire representation, something…
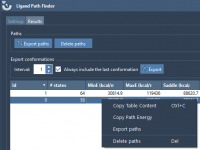
Molecular modelers often spend a great deal of time analyzing ligand unbinding pathways. Once paths are identified, it’s not uncommon to want to export the atomic coordinates corresponding to a subset of atoms moving along these paths—for example, to investigate…
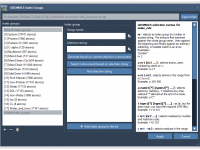
Creating custom index groups is a recurring need in molecular simulations with GROMACS. Whether you are working on protein-ligand complexes, membrane systems, or free energy calculations, being able to define and reuse specific atom or residue groups simplifies analysis, facilitates…
When modeling complex molecular systems, selecting exactly the right atoms, residues, or chains can get tedious. Even with powerful selection tools, the process often involves clicking through multiple menus or remembering specific selection syntaxes. This becomes especially frustrating when you…
When you’re working on a molecular movie or educational demo, it’s often useful to make certain molecules or parts of a system disappear for a few moments. Maybe you want to reveal an internal binding site, or highlight a dynamic…
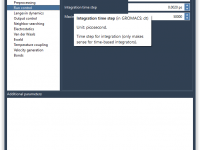
One of the common challenges molecular modelers face is adapting general-purpose parameter sets to the specific needs of their systems. Whether you’re simulating a novel peptide or working with non-standard residues, modifying molecular dynamics parameters (MDP parameters) is often essential.…
Constructing carbon nanotubes manually is a common yet time-consuming task in nanoscale modeling. Molecular modelers often spend significant time aligning repeating subunits, ensuring correct bonding distances, and stacking rings into tubular shapes — and that’s before even getting to energy…
Dragging atoms to explore structural rearrangements is a common task in molecular modeling, yet it often leads to frustration when the topology inconsistencies break your simulation or generate unphysical structures. If you’ve ever attempted to manually tweak a molecular structure…