Creating effective molecular animations often means more than just spinning a molecule or changing its size. Communicating a precise scientific idea might require revealing specific structures at key moments. But how do you do this without compromising visual clarity or…
When working with complex nanosystems, one of the biggest challenges scientists and engineers face is how to efficiently visualize and interact with molecular structures. It’s not just about seeing atoms and bonds — we need to understand higher-level organization, surface…
When loading molecular structures into SAMSON, you may find yourself dealing with scattered orientations or off-center models. This seemingly minor layout issue can become an obstacle when you want to build larger systems, compare multiple conformations, or prepare your scene…
Creating compelling visualizations in molecular modeling is often critical—whether to explain a docking mechanism, present a complex structure at a conference, or simply share your work with collaborators. But one challenge that holds many modelers back from producing dynamic molecular…
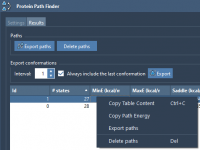
When studying protein conformational changes, modeling how a protein transitions from one conformation to another is just the beginning. Molecular modelers often need to analyze and share results: extract key conformations, view transition energies, or export trajectories for visualization and…
If you’re studying protein dynamics, a common challenge is identifying conformational pathways between two known states. Before diving into complex simulations or heavy computations, one crucial step must be done right: loading and preparing your protein’s conformational models correctly. In…
When working on complex molecular modeling projects, it’s often easy to lose track of elements in the document—especially when multiple cameras, lights, and visual nodes are involved. If you’ve ever found yourself trying to manually sift through camera nodes just…
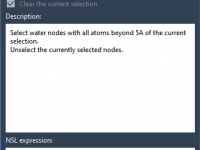
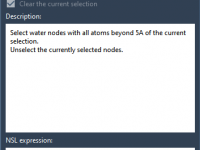
Water molecules in protein structures from the Protein Data Bank (PDB) can either be essential for function or relics from crystallographic data that don’t matter for your simulation. Knowing how to selectively remove undesired water molecules while preserving those in…
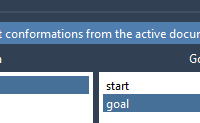
When modeling conformational transitions in proteins, ensuring that the start and goal structures are properly aligned and compatible is essential. If your starting and ending conformations are not set up correctly, most path-finding algorithms will either fail or return meaningless…
In molecular simulations, especially when preparing systems using GROMACS, it’s common to remove water molecules from the initial structure to avoid unwanted interactions or duplicate solvation. However, researchers often face a recurring dilemma: how can you quickly remove unnecessary crystal…