Determining the ideal transition pathway between two molecular states—say, before and after binding—is a core challenge in molecular modeling. Tools like the Parallel Nudged Elastic Band (P-NEB) method help researchers explore the potential energy landscape between structures. But if you’ve…
Molecular modeling often involves many small, consecutive actions—selecting atoms, building fragments, calculating energies, adjusting parameters. But what happens when you make an unintentional change several steps back and don’t notice until it’s too late? With SAMSON, you can breathe easier:…
It’s a common challenge for molecular modelers building scientific animations: how to present complex molecular mechanisms while ensuring the viewer’s attention is focused on the right parts—at the right time. Whether you’re illustrating ligand binding or signaling cascades, controlling when…
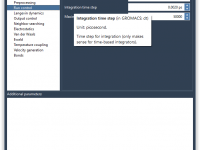
If you’ve ever tried to fine-tune your molecular dynamics simulations in GROMACS, you probably know how intimidating it can be to manage .mdp files manually. With models getting increasingly complex, juggling dozens of simulation parameters becomes a source of friction…
If you’ve ever wanted to adjust molecular geometries while watching energies and forces respond in real-time, SAMSON‘s interactive simulations offer an intuitive way to do so. But if you’ve never added a simulator before, the process can seem a bit…
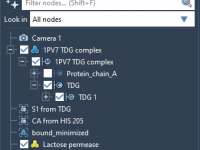
Anyone working with complex molecular models knows how quickly a project can become overwhelming. Once you have large protein structures, assemblies, or entire molecular systems with multiple chains, managing and navigating them effectively becomes a challenge. What if you’re only…
If you’ve ever prepared molecular animations for presentations, publications, or educational content, you know how time-consuming it can be to manually simulate atoms or molecular groups moving away from their binding sites. Whether you’re modeling a drug molecule departing from…
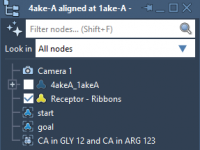
When modeling protein transitions — for instance, switching between open and closed conformations — a surprisingly common obstacle is just getting the input right. If your two conformations come in separate PDB files, you might scramble to align and manage…
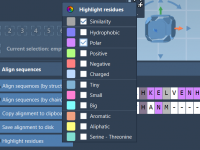
When comparing proteins across species, variants, or conditions, sequence alignment is often the first step. But for molecular modelers, aligning only sequences may not capture the impact of structural context — and that’s where SAMSON’s Protein Aligner becomes especially helpful.…
In molecular modeling, clarity is essential. When working with complex assemblies, it’s easy for molecular scenes to become visually overwhelming. SAMSON’s Node Specification Language (NSL) provides a simple way to control the visibility of elements using note attributes. This is…