Exploring Crystal Defects in Diamond with SAMSON’s Crystal Creator.
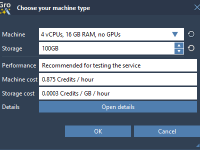
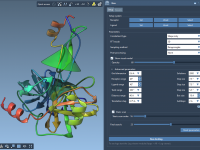
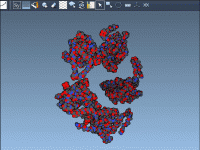
Molecular modelers often encounter challenges when attempting to simulate real-world crystals. Among these challenges, accounting for crystal defects—such as atom vacancies—is essential for accurately representing materials. The Crystal Creator Extension in SAMSON offers an intuitive way to explore and model…