When working with complex molecular models, clarity is key. Whether you’re preparing figures for publication, sharing a model with collaborators, or simply trying to declutter your workspace, the ability to selectively show or hide label nodes can make a significant…
When working with complex molecular trajectories, a common challenge is keeping your focus on a specific region of interest. Atoms may move, conformations may shift, and before you know it, your camera has lost track of the very part of…
A common challenge in molecular modeling is effectively communicating complex 3D structures and dynamic behaviors to both colleagues and broader audiences. Whether for presentations, publications, or teaching, clarity and visual engagement are key – and this is where animations shine.…
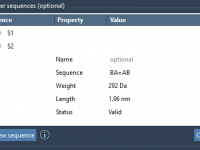
If you build custom polymers for molecular modeling, you’ve probably dealt with challenges when specifying complex monomer sequences. How do you know your defined sequence is valid? Have you correctly designated bond types between monomers? Are your naming conventions traceable?…
When it comes to creating molecular models that are both scientifically accurate and visually compelling, the choice of materials can make a massive difference. Whether you’re customizing metallic proteins, simulating glassy channels, or adding a glowing ligand for effect, the…
When working on complex molecular models in SAMSON, it’s common to use annotations or graphical labels to highlight specific regions, monitor simulation data, or organize your workspace. These label nodes are versatile, but as your models grow in size and…
When modeling complex molecular systems, isolating specific components—like molecular backbones based on their attributes—can save a considerable amount of time and help reduce visual clutter. If you’ve ever asked yourself, “How do I hide all backbones that are not selected?”…
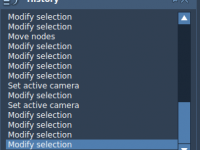
Whether you’re tweaking geometries, testing force field parameters, or building complex biomolecular assemblies, making changes efficiently and confidently is key. But with complexity comes a familiar concern among molecular modelers: what if you make a mistake and can’t go back?…
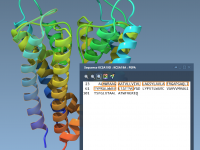
Anyone who models biomolecular systems knows how tedious it can be to manually pick specific residues in a 3D viewport. Whether you’re selecting residues for simulations, mutations, or analysis, navigating protein structures in 3D often leads to frustration—especially when chains…
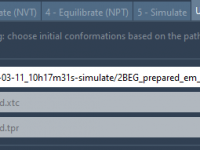
Running an Umbrella Sampling simulation can be time-consuming, especially when it comes to preparing the system correctly. One of the most critical — and often tedious — steps is choosing the right initial conformations. Missteps here can affect the reliability…