





Category: Uncategorized
When (and Why) to Minimize Ligands Before Docking
Smooth Transitions for Molecular Presentations: Using Background Interpolations in SAMSON
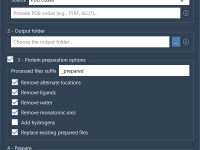
Working with Multiple PDB Files? Here’s How to Clean Them All at Once
Filter and Find Node Groups in Your Molecular Models with Precision
A Simple Way to Control Fragment Orientation in Your Molecular Models
A Smoother Perspective: Vertical Camera Transitions with Pedestal Animation in SAMSON
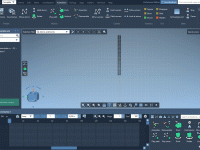
When preparing molecular animations for presentations, publications, or educational content, achieving smooth and controlled camera movements is essential. A common challenge many molecular modelers face is aligning camera motion with the structure’s spatial dimensions—especially in the vertical direction. This is…
Managing Multiple Molecules? Use SAMSON Documents and Folders Effectively
Quickly Hide or Filter Notes in SAMSON with Attribute Flags
Quick Groups in SAMSON: Save and Recall Your Molecular Selections Instantly

In molecular modeling, repetitive selection of the same atoms, residues, or fragments can quickly become tedious—especially when dealing with complex biomolecular systems. Whether you’re analyzing ligand binding sites, highlighting functional groups, or toggling between different molecular regions, having to re-select…
When (and Why) to Minimize Ligands Before Docking
Smooth Transitions for Molecular Presentations: Using Background Interpolations in SAMSON
Working with Multiple PDB Files? Here’s How to Clean Them All at Once
Filter and Find Node Groups in Your Molecular Models with Precision
A Simple Way to Control Fragment Orientation in Your Molecular Models
A Smoother Perspective: Vertical Camera Transitions with Pedestal Animation in SAMSON
When preparing molecular animations for presentations, publications, or educational content, achieving smooth and controlled camera movements is essential. A common challenge many molecular modelers face is aligning camera motion with the structure’s spatial dimensions—especially in the vertical direction. This is…
Managing Multiple Molecules? Use SAMSON Documents and Folders Effectively
Quickly Hide or Filter Notes in SAMSON with Attribute Flags
Quick Groups in SAMSON: Save and Recall Your Molecular Selections Instantly
In molecular modeling, repetitive selection of the same atoms, residues, or fragments can quickly become tedious—especially when dealing with complex biomolecular systems. Whether you’re analyzing ligand binding sites, highlighting functional groups, or toggling between different molecular regions, having to re-select…