Category: Uncategorized
Syncing Trajectories with Ease: Using the Play Path Animation in SAMSON
One of the recurring pains in molecular modeling is the clear and synchronized visualization of structural transitions—whether you’re browsing a molecular dynamics trajectory, comparing different conformers, or showing simulation results in a presentation. When you need to animate changes seamlessly…
Using the Flash Animation to Emphasize Molecular Events Without Overcomplicating the Scene
Speeding up Nanotube Design with Pattern Editors in SAMSON
For molecular modelers working on nanostructures, building carbon nanotubes and similar architectures atom by atom is time-consuming and error-prone. Small misalignments between atoms can create structurally incoherent models and increase the time required for cleanup and minimization. If your work…
Quickly Filter Molecular Side Chains by Atom Type in SAMSON
When dealing with large biomolecular models, pinpointing specific side chains with certain atomic compositions can be tedious and time-consuming. SAMSON’s Node Specification Language (NSL) provides a concise way to filter and select these molecular entities efficiently. Whether you’re preparing systems…
Why Protein Cleanup Matters Before Structural Interpolation
From Steel to Glass: Fine-Tuning Molecular Materials with Cycles in SAMSON
When Molecules Fade Away: Using ‘Conceal atoms’ to Focus Your Story
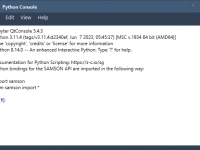
Python Scripting in SAMSON: Automate, Simulate, Repeat
How to Select Charged, Polar, or Hydrophobic Residues in Seconds
Syncing Trajectories with Ease: Using the Play Path Animation in SAMSON
One of the recurring pains in molecular modeling is the clear and synchronized visualization of structural transitions—whether you’re browsing a molecular dynamics trajectory, comparing different conformers, or showing simulation results in a presentation. When you need to animate changes seamlessly…
Using the Flash Animation to Emphasize Molecular Events Without Overcomplicating the Scene
Speeding up Nanotube Design with Pattern Editors in SAMSON
For molecular modelers working on nanostructures, building carbon nanotubes and similar architectures atom by atom is time-consuming and error-prone. Small misalignments between atoms can create structurally incoherent models and increase the time required for cleanup and minimization. If your work…
Quickly Filter Molecular Side Chains by Atom Type in SAMSON
When dealing with large biomolecular models, pinpointing specific side chains with certain atomic compositions can be tedious and time-consuming. SAMSON’s Node Specification Language (NSL) provides a concise way to filter and select these molecular entities efficiently. Whether you’re preparing systems…