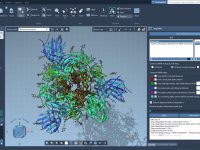
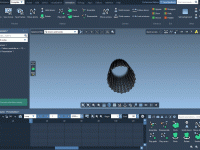
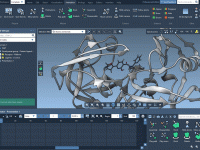
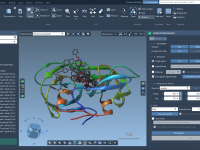
One of the surprisingly common pains molecular modelers face is interpolating the motion between two conformational states of a macromolecular complex when those structures differ in the number of atoms or residues. Even small differences can impede trajectory generation, especially…
Running a molecular dynamics (MD) simulation is a critical step in exploring the dynamic behavior of molecular systems like proteins, membranes, or drug candidates. But once the simulation ends, the next challenge can feel just as important — making sense…
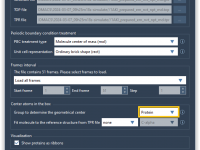
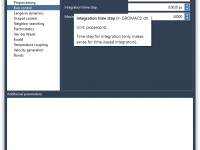
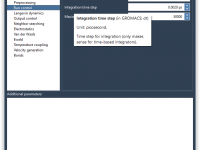
For many molecular modelers using GROMACS, customizing simulations often means diving into .mdp files and adjusting text-based parameters manually. While this gives flexibility, it can also be error-prone and lacks visual guidance. What if you could tweak your simulation settings…
When creating molecular animations, there’s often a fine balance between aesthetic fluidity and scientific clarity. A small but recurring challenge arises when a researcher wants to zoom into a particular region of a molecular system without shifting the overall perspective…
Working with complex molecular structures can be frustrating when you’re trying to locate specific segments or unsure whether a segment meets certain requirements. Whether analyzing a model with thousands of atoms or attempting to filter specific residues, identifying the right…
One common challenge in molecular modeling and presentation is managing visual clarity when dealing with complex systems. It’s easy for key elements to get lost in a mess of atoms and bonds, especially in animated sequences where the molecular structure…
When working with large molecular systems, searching for specific atom types—aromatic carbons, water oxygens, charged residues—can feel like finding a needle in a haystack. Whether you’re setting up a simulation, preparing a molecular surface analysis, or generating properties to color…
For many molecular modelers, switching between development environments, simulation tools, and analysis platforms is a regular part of the workflow. This fragmentation can be time-consuming and error-prone, especially when adapting or reusing previously developed code. But what if your trusted…
If you’re a molecular modeler using GROMACS through the SAMSON platform, you may have tried to customize molecular dynamics parameters for different simulation steps—only to discover later that some of your settings were not applied during the run. This can…
When preparing molecular animations, researchers often face a common challenge: how to focus attention on meaningful movement without unnecessary distractions. If everything in the scene moves, it can become harder to understand what matters. Sometimes, it’s crucial to anchor part…