





When setting up molecular dynamics simulations, especially in biological or materials science contexts, one often faces an overwhelming number of choices. One such choice that significantly impacts both simulation accuracy and performance is the shape of your unit cell used with periodic boundary conditions. While this might feel like a minor technicality, it can translate into substantial computational savings and influence the reliability of your simulations.
The GROMACS Wizard in SAMSON offers several unit cell options when preparing a molecular system. So, which one should you choose? And what difference does it make?
Why care about unit cell shape?
GROMACS uses periodic boundary conditions (PBCs), essentially placing your system in a repeating box that mimics an infinite environment. This is essential for simulating bulk properties or solvated systems without edge effects. However, the shape of this box — or unit cell — can affect how many solvent molecules you need, how particles interact across boundaries, and ultimately, how fast and accurate your results are.
Overview of Common Unit Cell Shapes
The GROMACS Wizard supports the following unit cell geometries:
| Unit cell shape | Representation |
|---|---|
| Cubic |  |
| Orthorhombic |  |
| Triclinic |  |
| Rhombic dodecahedron |  |
| Truncated octahedron |  |
What’s the most efficient option for solvated macromolecules?
When simulating spherical or globular biomolecules in solution, the rhombic dodecahedron and truncated octahedron cells can save significant computational time. These shapes more closely conform to a sphere than a cube does. As a result, you require fewer water molecules to surround your solute with a given minimum image distance — reducing simulation box volume and computation cost.
In fact, a rhombic dodecahedron has only 71% of the volume of the equivalent cubic box. That’s a potential 29% reduction in CPU time for similar solvation conditions, making it an excellent choice for proteins, nucleic acids, or other approximately spherical structures.
How to choose and fit your box in SAMSON
GROMACS Wizard allows you to choose both your unit cell type and whether to set the box using:
- Box lengths – You define the exact dimensions, and can reposition the solute within the box. Recommended when you want consistent box sizes across multiple conformations (e.g., a batch of simulations).
- Solute-box distance – You define how far from the solute the box boundary should start. Ideal for systems undergoing large conformational changes, ensuring minimum image convention is preserved for each conformation individually.
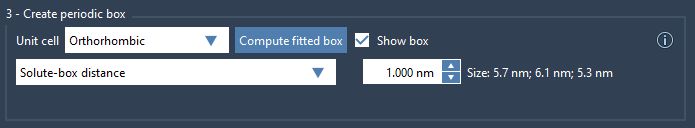
Whichever method you pick, aim for at least 1.0 nm distance between the solute and the box boundary. This ensures forces are calculated correctly and no solute sees its periodic image. This recommendation is consistent with most cutoff schemes used in molecular dynamics simulations.
A note about visualization and trajectory import
GROMACS stores simulation boxes as brick-shaped for performance reasons. When analyzing trajectories in SAMSON, the platform attempts to deduce the original unit cell shape during import. However, you can still modify this choice manually if needed in the importer dialog — useful when working with non-cubic cells like the rhombic dodecahedron.
Choosing the right unit cell is not just about technical preference — it’s about smarter simulations. Understanding the options and their implications helps you make informed decisions for both single simulations and entire workflows.
To learn more, visit the official SAMSON documentation page on periodic boundary conditions.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON from https://www.samson-connect.net.