





Preparing input files for free energy calculations can often be tedious, requiring custom scripts to extract atomic coordinates along a reaction path. Fortunately, SAMSON’s Export Along Paths extension streamlines this task through a user-friendly interface that allows you to export atom trajectories along predefined paths — useful when working with ligand unbinding simulations or enhanced sampling methods.
In this post, we’ll explore how to select and export the trajectory of a ligand (such as TDG in a lactose permease system) along a reaction path computed in SAMSON. This is particularly helpful if you’re calculating a potential of mean force using umbrella sampling or other free energy approaches 🎯.
Why extract only one molecule’s path?
Molecular dynamics generates a lot of data. But for specific applications like reaction coordinate-based methods, you typically care about just a subset of that data — for example, the behavior of a ligand as it exits a protein tunnel. Exporting only the ligand trajectory keeps things simple and reduces data processing time later in tools like GROMACS or AMBER.
Step-by-step: Exporting a subset of atoms along a path
Let’s say you’ve already used the Ligand Path Finder to define unbinding paths. Here’s how to export a specific set of atoms like your ligand:
- Launch the Export Along Paths app in SAMSON: go to
Home > Apps > All > Export Along Paths. - Expand the Advanced panel in the app interface.
- Select the ligand (e.g.,
TDG) in the Document View.
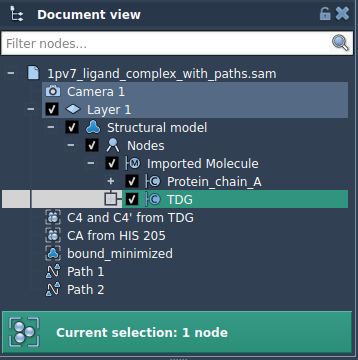
- Click Add in the Export Along Paths app to define this selection as a model for export. This makes it easy to track what gets exported and allows for multiple selections.
You will now see the selected ligand in a list, and you can:
- Rename it for clarity.
- Add other atom sets the same way.
Once ready:
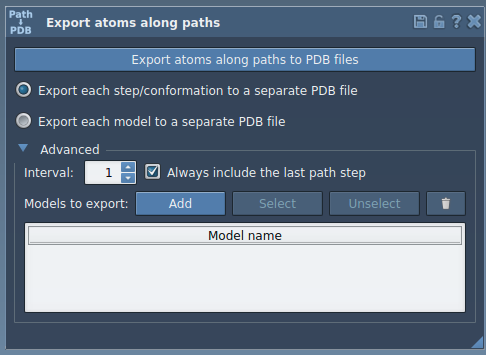
- Choose whether to export one PDB with all frames or one file per frame.
- Select the relevant path(s) in the Document View.
- Click Export atoms along paths to PDB files.
Use cases
This approach is especially useful when generating input files for methods that require reaction coordinate definitions, such as:
- Umbrella sampling
- Metadynamics
- Free energy perturbation studies where intermediate states are needed
You can also export multiple components (e.g., ligand and binding pocket residues) separately and reassemble them downstream, leading to cleaner workflows 😌.
Conclusion
By selecting and exporting trajectories for just the atoms you care about, you can save time and reduce error in your analyses. SAMSON makes this practical with minimal setup and a graphical interface.
Learn more in the full documentation
SAMSON and all SAMSON Extensions are free for non-commercial use. You can get SAMSON here.