





For molecular modelers, accurately finding minimum energy paths between molecular conformations is a vital aspect of understanding reactions, binding mechanisms, and structural transitions. Yet, bridging the gap between initial and final states swiftly and precisely can often be a challenge. Enter the Parallel Nudged Elastic Band (P-NEB) app: a tool integrated into SAMSON to optimize these paths with ease and efficiency.
The P-NEB app builds on the established Nudged Elastic Band (NEB) method, which achieves a clear solution to this problem by identifying saddle points and minimum energy pathways between two conformational states. Importantly, it incorporates constrained optimization, refining intermediary images of the transition using spring forces while maintaining uniform spacing. By doing this in parallel, as its name suggests, P-NEB accelerates computation without sacrificing precision.
Why Use P-NEB for Molecular Pathways?
Whether you’re investigating how ligands unbind from proteins or observing shifts in molecular architectures, P-NEB offers a structured pathway for tackling complex intermediary states. Transition paths often consist of numerous images that need to be optimized. P-NEB reduces this workload by leveraging multi-threading capabilities, allowing each conformation to be refined in parallel. This efficiency enables you to scale your analysis for larger systems or examine multiple transition paths simultaneously.
Getting Started with P-NEB
To begin, ensure you have the required SAMSON extensions:
- The P-NEB Extension
- The FIRE (Fast Inertial Relaxation Engine) state updater
You can follow this up by loading one of the sample molecular models available in SAMSON. For instance, the Zinc ligand trajectory or the Lactose permease system (1PV7) are excellent places to start exploring. These samples are accessible directly on Home > Download in SAMSON’s interface.
How to Use P-NEB for Path Optimization
Once your system is set up, activating the P-NEB app is straightforward. Head to Home > Apps > All > P-NEB, or simply find it through the universal search box in SAMSON.
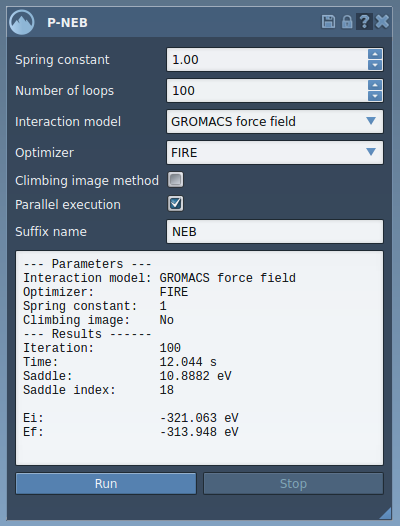
The P-NEB interface allows you to configure several key parameters for the optimization:
- Spring constant: Set the tension for the spring forces responsible for constraining the optimization.
- Interaction model: Choose from available force fields like the “Universal Force Field (UFF)” for energy computation.
- Parallel Execution: Enable this for multi-threaded optimizations, which speeds up the process.
- Climbing Image Method: Leave this unchecked in initial runs, but feel free to test it later to locate precise saddle points.
To optimize, either select an existing path or create one by combining individual conformations (Edit > Conformation) in the Document view. Simply click “Run” in the P-NEB interface and let the app handle the computation. The resulting optimized path will appear in the Document view, allowing for further examination and animations to validate your results.
A Time-Saving Tip
If you’re working with a system that has two known relaxed states, creating a path via linear interpolation or tools like SAMSON’s Ligand Path Finder can save valuable time. Directly applying P-NEB to a pre-defined path tends to outperform application to a set of independent conformations in terms of efficiency.
Wrapping Up
The P-NEB app is an indispensable addition to any molecular modeler’s toolkit, aiding in the study of structural dynamics with remarkable efficiency. By taking advantage of parallel computations, it enables researchers to navigate complex energy landscapes without compromising precision. To dive deeper and explore further use cases, visit the official documentation page.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. To get started, visit SAMSON Connect.