





When you are studying large macromolecular systems, one notoriously difficult task is to simulate plausible transitions between two known conformations. Whether you’re investigating protein folding, loop dynamics, or viral mechanisms, modeling intermediate structures between an open and closed state can be both time-consuming and error-prone.
If you work with proteins like the SARS-CoV-2 spike, you might have the structures for the closed and open states. But producing a meaningful trajectory between those two can be tricky, especially when the conformations differ in their residue count or local structure. That’s where SAMSON can save time — by combining automated trajectory interpolation with refinement steps.
Interpolating Transition Paths Safely
Instead of manually crafting motions or using full-blown molecular dynamics — often infeasible at this scale — you can compute a structural transition using a mix of:
- ARAP (As-Rigid-As-Possible) interpolation to generate an initial path.
- P-NEB (Parallel Nudged Elastic Band) to refine it with physically realistic transitions.
In the case of the SARS-CoV-2 spike protein, researchers used the PDB entries 6VXX (closed state) and 6VYB (open state) as inputs. However, because these contain different numbers of residues, a naive interpolation would break. Here’s how they solved it:
- Ensure correct bond orders in sugars using a Python script.
- Add hydrogens and perform structure minimization.
- Use ARAP to interpolate from the open to closed conformation (producing a smooth path).
- Extract a conformation from the ARAP trajectory that structurally matches the open state (since ARAP paths stay in the same structure).
- Regenerate a new ARAP path using that intermediate conformation as the goal.
- Refine the path using the P-NEB module to produce a physically more plausible transition.
Why It Matters
This multi-step approach allows you to generate realistic intermediate conformations quickly, even when your start and end structures differ in residue counts or topologies. It’s particularly useful when experimental data is sparse and speculative motions are hazardous. This method helps you:
- Visualize functional mechanisms in proteins like viral spikes or enzyme loops.
- Generate frames for docking simulations or for training machine learning models.
- Share hypothesis about conformational transitions supported by plausible intermediates.
Animation Example: The Spike Transition in Action
The transition from the down (closed) to up (open) state of the SARS-CoV-2 spike reveals how one RBD arm rises to engage with the human ACE2 receptor. This animation, computed with ARAP and refined with P-NEB, brings that transition to life:
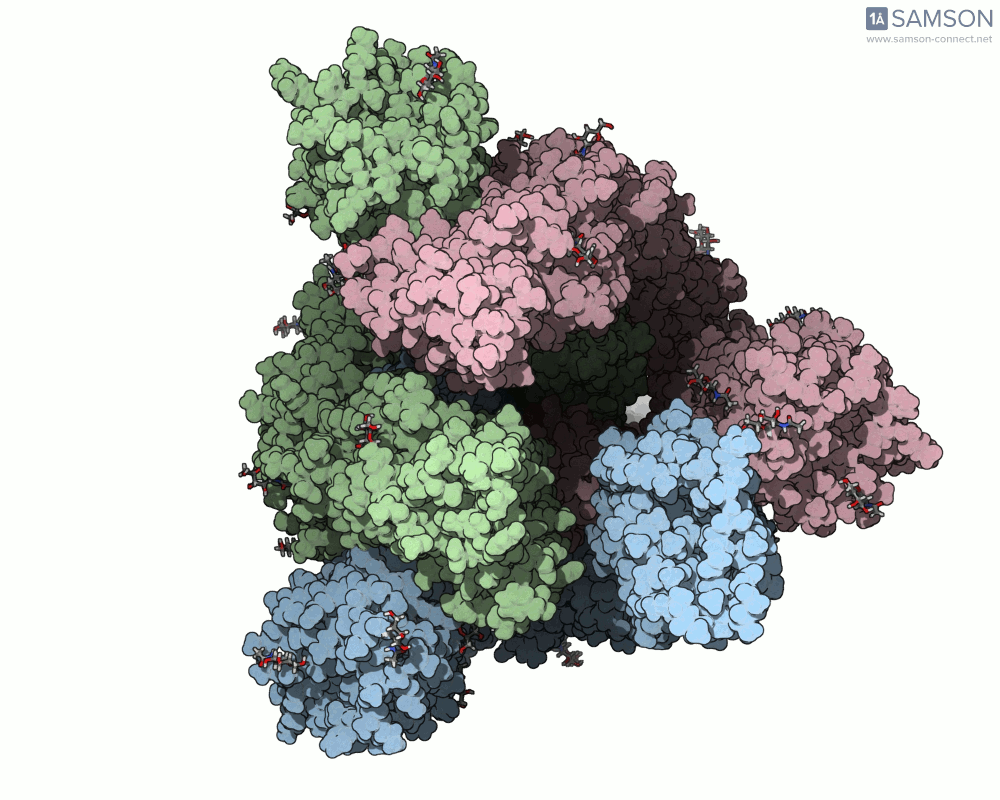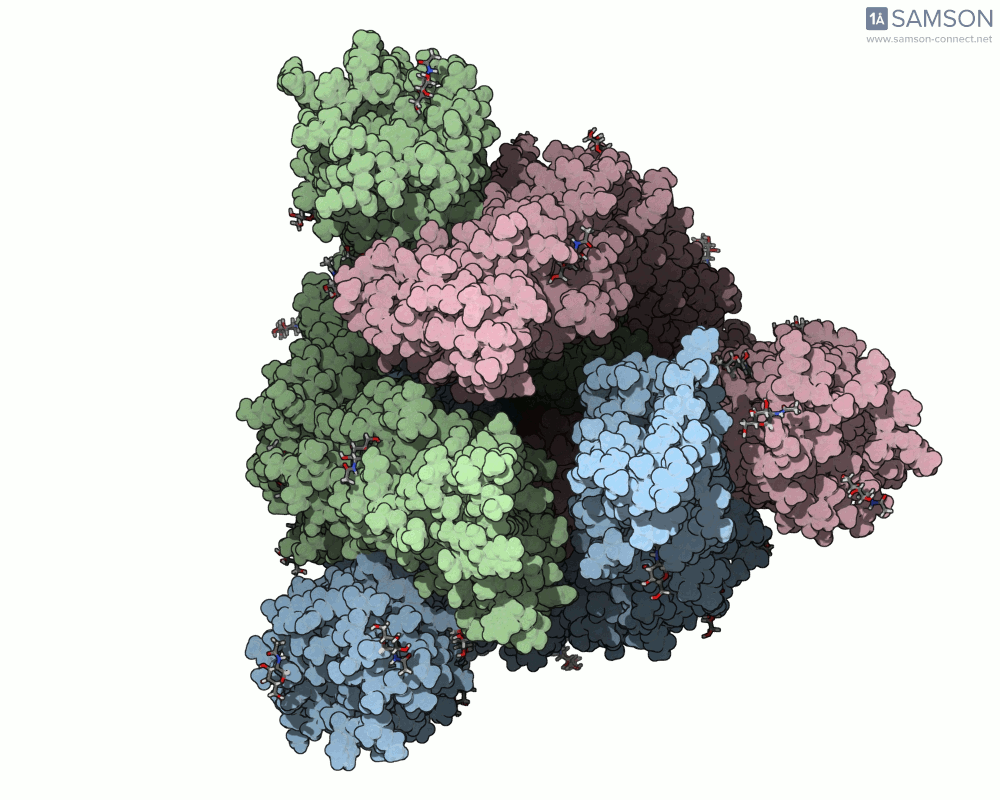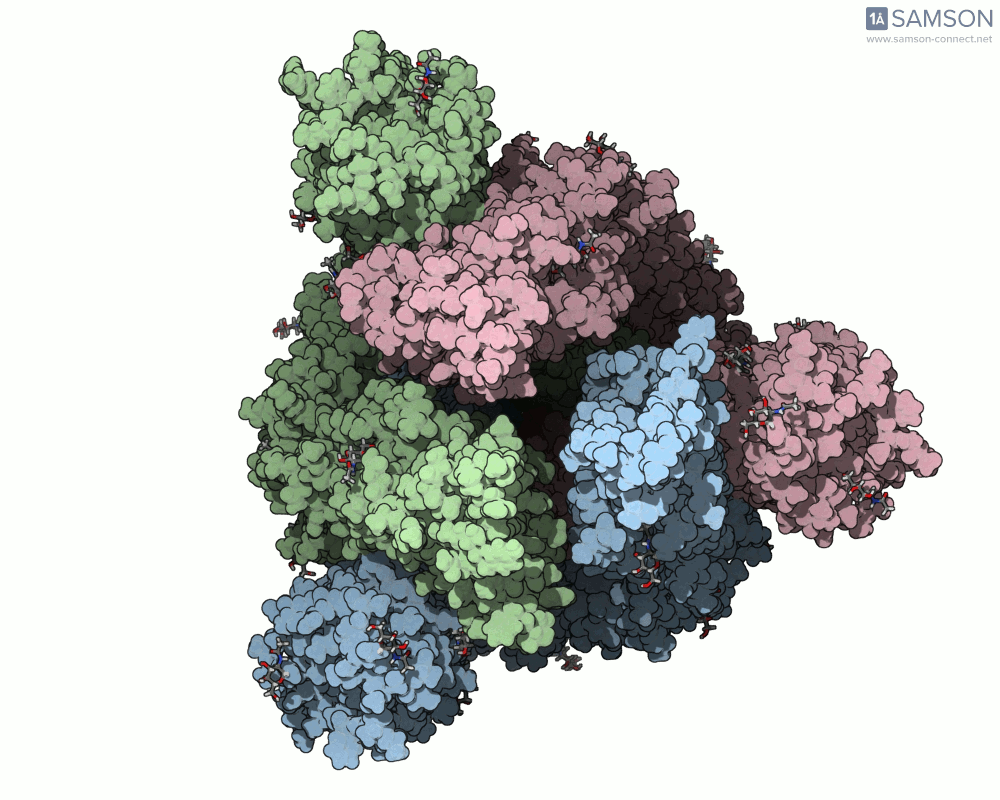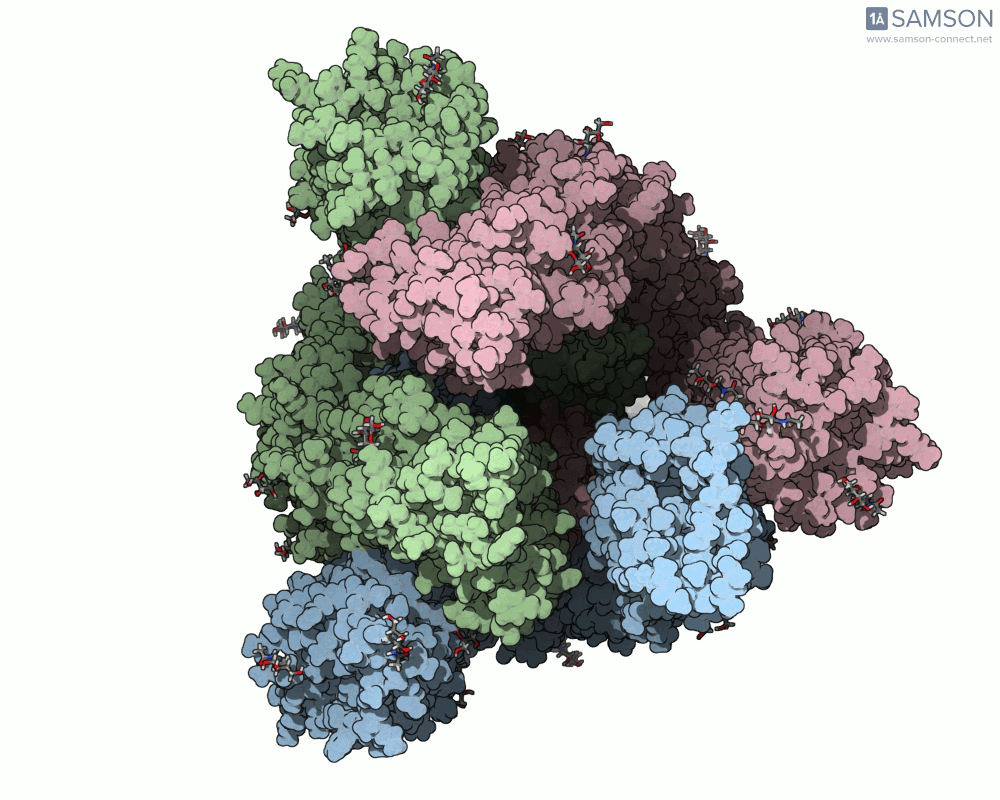
Useful Output Files
To make your work even easier, the team has shared downloadable trajectory files:
This offers a reproducible pipeline for others to learn from or adapt to their own systems, such as other viral entry proteins or flexible domains in large macromolecules.
To explore the full tutorial and download modules, visit the official SAMSON documentation page.
SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON here.