





Setting up molecular simulations can often be a daunting task, especially for molecular modelers who aim to balance accuracy with ease of use. Fortunately, SAMSON’s integration of the Universal Force Field (UFF) helps streamline this process significantly. This blog post provides a step-by-step guide on how to set up UFF simulations in SAMSON effectively. Whether you are tweaking molecular structures or preparing complex systems, this guide covers the essentials to get started seamlessly and perform meaningful calculations.
Why Use the Universal Force Field?
The Universal Force Field (UFF) provides a versatile molecular mechanics model that spans the periodic table, making it suitable for a wide range of molecular systems. Its applications extend to bond computation, atom typing, and energy forecasting in molecular systems. SAMSON’s implementation of UFF also includes an automatic perception scheme, which simplifies workflows by computing bonds, bond orders, and atom types directly from atom positions. If you need rapid setup for reliable simulations, UFF offers a strong foundation.
Step-by-Step Guide to UFF Setup
Follow these simple steps to bring the power of UFF into your molecular simulations:
- Load Your Molecular System: Open a molecular document in SAMSON that requires simulation using UFF.
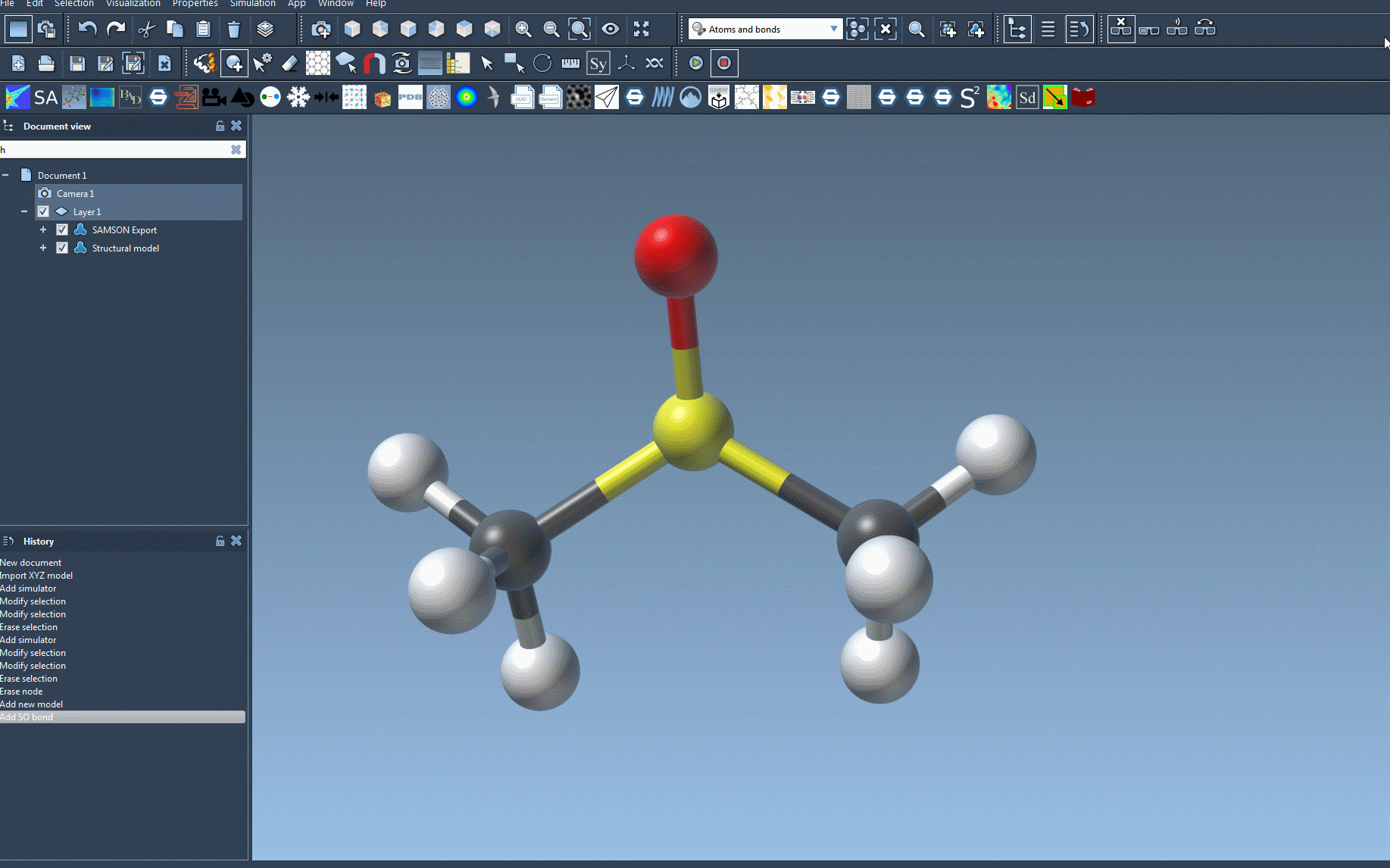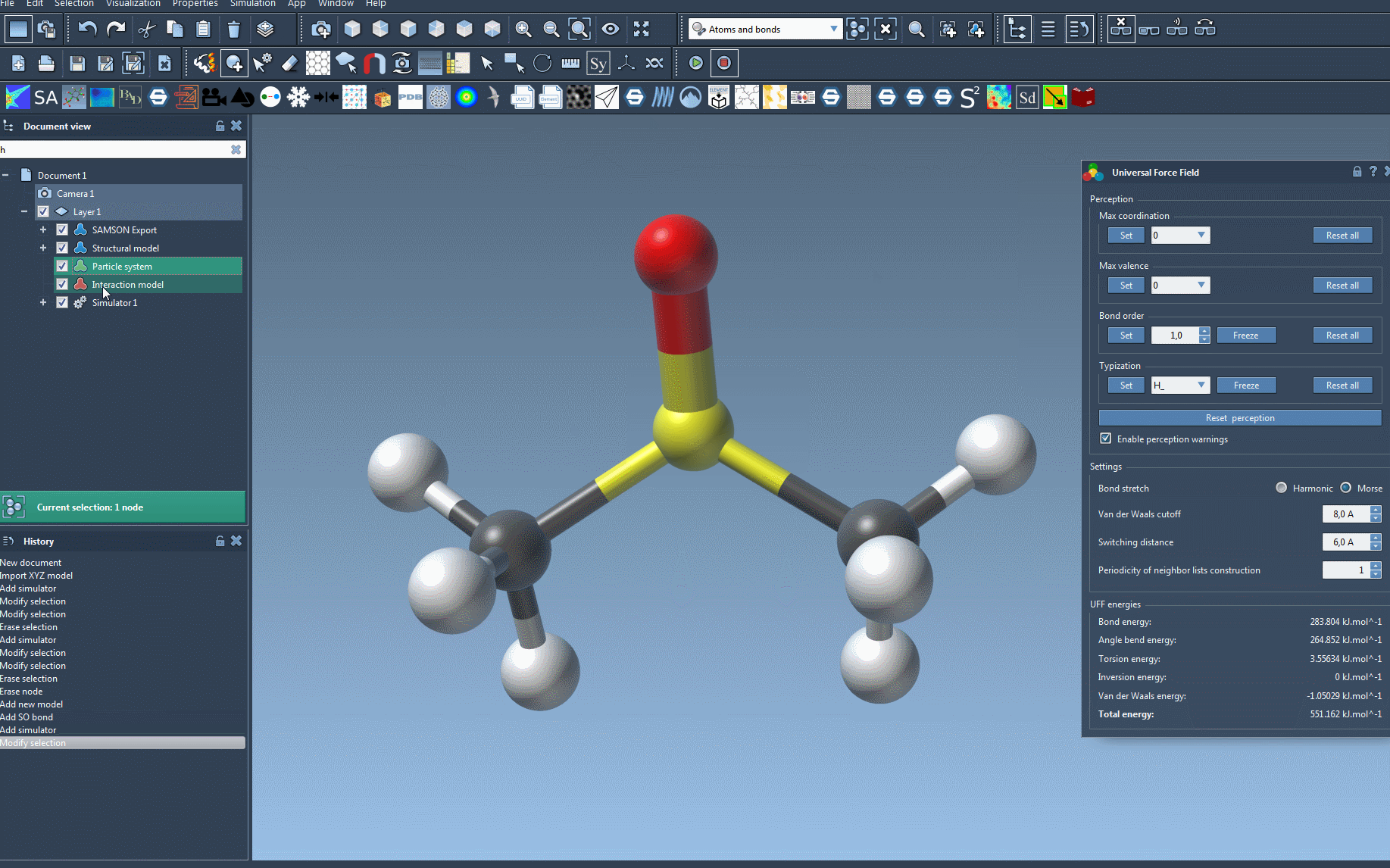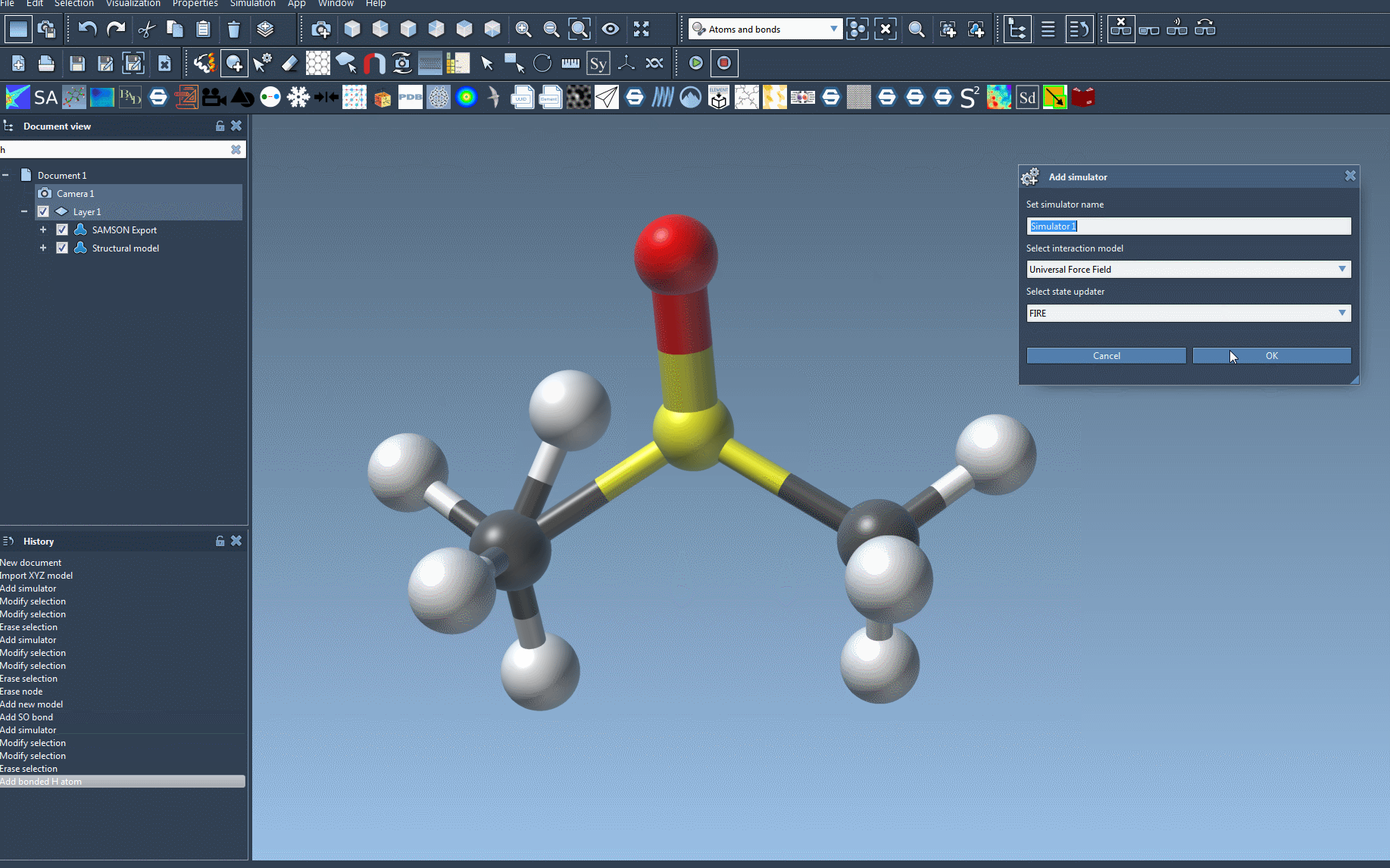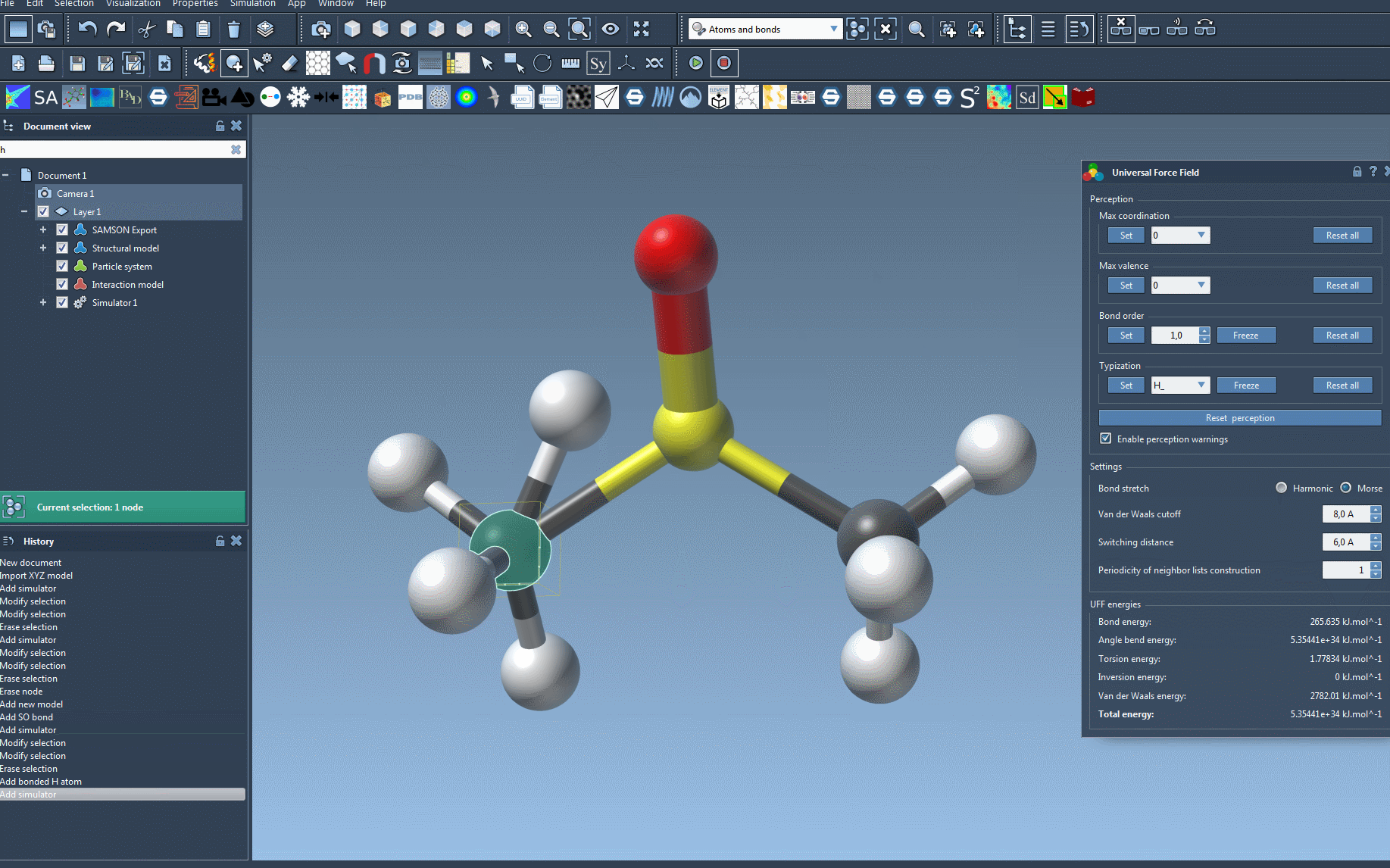
- Add the UFF Simulator:
- Navigate to
Edit > Simulate > Add simulator. Alternatively, use the shortcut: - Ctrl+Shift+M (Windows/Linux) or Cmd+Shift+M (Mac).
- Navigate to
- Select Interaction Model: In the simulator options list, choose Universal Force Field as the interaction model.
- Choose a State Updater: You can utilize state updaters like the FIRE minimizer for an optimized simulation workflow.
- Confirm Setup: Click the OK button to proceed.
Once these steps are complete, the UFF setup window will appear, allowing further customizations.
Customizing the Setup
You can streamline UFF setup efficiently depending on your molecular system’s characteristics:
- Using Existing Bonds: Check the Use existing bonds option if your molecular model already defines bonds clearly. Otherwise, UFF will calculate bonds from atom types and positions.
- Automatic Initialization: Once confirmed, SAMSON will handle computations for covalent bonds, bond orders, and atom types.
During initialization, SAMSON will alert you if any inconsistencies are detected in your molecular system, giving you the opportunity to correct them before moving forward.
Key Advantages of SAMSON’s UFF Tool
As you experiment with UFF in SAMSON, you will appreciate these benefits:
- Interactive atom manipulation: Move atoms and observe energy and structural changes dynamically.
- Customizable parameters: Toggle between harmonic and Morse bond stretch interactions, adjust van der Waals cutoff distances, and more.
- Streamlined workflows: With automatic perception of molecular structures, you can prepare simulations faster while maintaining accuracy.
Need additional guidance or want to dive deeper into UFF functionality? Explore the detailed documentation at UFF Setup Documentation.
Conclusion
By following the above steps, molecular modelers can efficiently set up Universal Force Field systems in SAMSON, eliminating bottlenecks while ensuring simulation precision. The UFF’s interactive and automated features make it an indispensable tool for professionals and beginners alike.
For a comprehensive breakdown of UFF’s full features and additional tutorials, visit the official documentation page at SAMSON’s UFF Tutorial.
Note: SAMSON and all SAMSON Extensions are free for non-commercial use. You can download SAMSON from here.