





A Quick Guide to Node Types in SAMSON
Streamlining Polymer Design with Sequence Registration
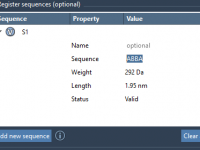
One of the challenges in molecular modeling and materials design is creating complex, custom polymers with repeating sequence patterns. Whether you’re constructing synthetic polymers for material property predictions or drug-polymer conjugates, manually assembling these structures can be tedious and error-prone.…
Mastering Ligand Pathways with SAMSON’s Ligand Path Finder.
Streamline File Import for Molecular Modeling in SAMSON
Streamlining Molecular Architecture with SAMSON’s Pattern Builders.
Effortlessly Organize Molecular Presentations with the Stop Animation
Effortlessly Moving Your Camera in Molecular Presentations
Proximity Operators in NSL: Streamlining Molecular Selections in SAMSON
Molecular modelers often face the challenge of finding specific nodes—atoms, residues, or structural groups—that meet particular spatial criteria within large and complex molecular structures. This task can be time-consuming and error-prone. Fortunately, SAMSON’s Node Specification Language (NSL) offers a powerful…