Mastering Simulate Animations for Constrained Molecular Simulations.
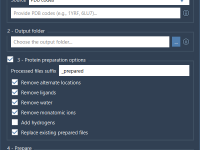
One recurring challenge for molecular modelers is achieving precise control during simulations, especially when simulating complex nanosystems where certain constraints or specific movements need to be maintained. Enter the Simulate animation in SAMSON, a powerful feature that facilitates constrained molecular…