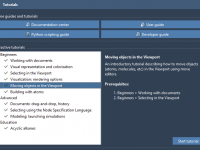
Master Molecular Modeling Workflows with SAMSON’s Interactive Tutorials
Safely Exploring Molecular Structures with SAMSON’s Undo and Redo Feature
Mastering Advanced Selections With Node Specification Language (NSL)
Achieving Faster Geometry Optimization with the FIRE Minimizer.

Mastering Node Visibility: Flash Animation in SAMSON
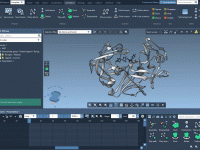
Simplify Molecular Modeling with Symmetry Detection in SAMSON

Simplifying Molecular Modeling Workflows with SAMSON Apps
For molecular modelers, managing complex workflows can be both time-consuming and challenging. Whether it is connecting to external tools, integrating computational services, or expanding functionalities with dedicated software, streamlining these tasks is vital to boost productivity. This is where SAMSON…