Exploring the SAMSON Extensions Marketplace

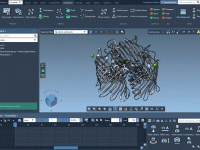
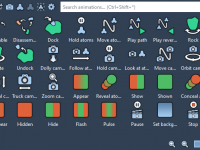
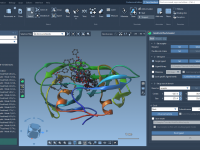
Molecular modeling often requires specialized tools for different tasks, from simulating molecular dynamics to analyzing complex structures. However, finding and integrating these tools can sometimes be challenging for researchers and developers. This is where the SAMSON Extensions Marketplace comes in…