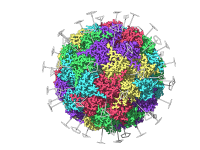
Tips for Assembling Molecular Fragments in SAMSON Platform
Molecular modelers often face the challenge of efficiently building complex molecular systems. Whether working on proteins, nanoparticles, or small molecules, creating accurate molecular structures quickly and intuitively can be demanding. SAMSON, the integrative molecular design platform, offers a powerful set…